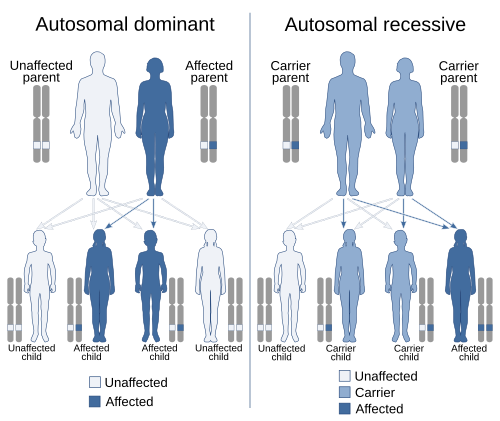
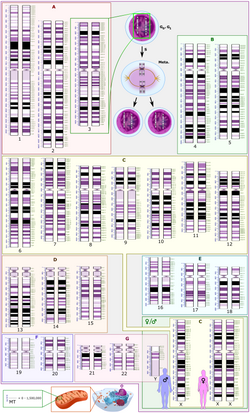
Medical genetics is the branch of medicine that involves the diagnosis and management of hereditary disorders. Medical genetics differs from human genetics in that human genetics is a field of scientific research that may or may not apply to medicine, while medical genetics refers to the application of genetics to medical care. For example, research on the causes and inheritance of genetic disorders would be considered within both human genetics and medical genetics, while the diagnosis, management, and counselling people with genetic disorders would be considered part of medical genetics.
In contrast, the study of typically non-medical phenotypes such as the genetics of eye color would be considered part of human genetics, but not necessarily relevant to medical genetics (except in situations such as albinism). Genetic medicine is a newer term for medical genetics and incorporates areas such as gene therapy, personalized medicine, and the rapidly emerging new medical specialty, predictive medicine.
In some ways, many of the individual fields within medical genetics are hybrids between clinical care and research. This is due in part to recent advances in science and technology (for example, see the Human Genome Project) that have enabled an unprecedented understanding of genetic disorders.[citation needed]
Individuals seeking acceptance into clinical genetics training programs must hold an MD, or in some countries, an MB ChB or MB BS degree. These qualifications ensure that trainees have the foundational medical knowledge required to specialize in Medical Genetics. The optimal training program involves a total of five years: one year of general medical training (the "common trunk", often covering fields such as general practice, pediatrics, obstetrics and gynecology, neurology, psychiatry, and internal medicine) followed by four years of specialized training in Medical Genetics. This specialized training should include at least two years of clinical patient care and at least six months in genetic laboratory diagnostics. Trainees' progress is evaluated through a structured program that begins with observation and progresses to independent practice under supervision, culminating in the ability to manage complex cases independently.[citation needed]
Final certification involves a comprehensive assessment, which may include national examinations or the European Certificate in Medical Genetics and Genomics (ECMGG). This certificate serves as a benchmark for high standards in the specialty across Europe and is increasingly recognized by various national regulatory authorities.[citation needed]
In the United States, physicians who practice clinical genetics are accredited by the American Board of Medical Genetics and Genomics (ABMGG).[1] In order to become a board-certified practitioner of Clinical Genetics, a physician must complete a minimum of 24 months of training in a program accredited by the ABMGG. Individuals seeking acceptance into clinical genetics training programs must hold an M.D. or D.O. degree (or their equivalent) and have completed a minimum of 12 months of training in an ACGME-accredited residency program in internal medicine, pediatrics, obstetrics and gynecology, or other medical specialty.[2]
Mitochondrial genetics concerns the diagnosis and management of mitochondrial disorders, which have a molecular basis but often result in biochemical abnormalities due to deficient energy production.[citation needed]
Genetic counseling is the process of providing information about genetic conditions, diagnostic testing, and risks in other family members, within the framework of nondirective counseling. Genetic counselors are non-physician members of the medical genetics team who specialize in family risk assessment and counseling of patients regarding genetic disorders. The precise role of the genetic counselor varies somewhat depending on the disorder. When working alongside geneticists, genetic counselors normally specialize in pediatric genetics which focuses on developmental abnormalities present in newborns, infants or children. The major goal of pediatric counseling is attempting to explain the genetic basis behind the child's developmental concerns in a compassionate and articulated manner that allows the potentially distressed or frustrated parents to easily understand the information. As well, genetic counselors normally take a family pedigree, which summarizes the medical history of the patient's family. This then aids the clinical geneticist in the differential diagnosis process and help determine which further steps should be taken to help the patient.[4]
History
Although genetics has its roots back in the 19th century with the work of the Bohemian monk Gregor Mendel and other pioneering scientists, human genetics emerged later. It started to develop, albeit slowly, during the first half of the 20th century. Mendelian (single-gene) inheritance was studied in a number of important disorders such as albinism, brachydactyly (short fingers and toes), and hemophilia. Mathematical approaches were also devised and applied to human genetics. Population genetics was created.[citation needed]
Medical genetics was a late developer, emerging largely after the close of World War II (1945) when the eugenics movement had fallen into disrepute.[5] The Nazi misuse of eugenics sounded its death knell.[6] Shorn of eugenics, a scientific approach could be used and was applied to human and medical genetics. Medical genetics saw an increasingly rapid rise in the second half of the 20th century and continues in the 21st century.[citation needed]
Current practice
The clinical setting in which patients are evaluated determines the scope of practice, diagnostic, and therapeutic interventions. For the purposes of general discussion, the typical encounters between patients and genetic practitioners may involve:[citation needed]
Referral to an out-patient genetics clinic (pediatric, adult, or combined) or an in-hospital consultation, most often for diagnostic evaluation.
Referral for counseling in a prenatal genetics clinic to discuss risks to the pregnancy (advanced maternal age, teratogen exposure, family history of a genetic disease), test results (abnormal maternal serum screen, abnormal ultrasound), and/or options for prenatal diagnosis (typically non-invasive prenatal screening, diagnostic amniocentesis or chorionic villus sampling).
Multidisciplinary specialty clinics that include a clinical geneticist or genetic counselor (cancer genetics, cardiovascular genetics, craniofacial or cleft lip/palate, hearing loss clinics, muscular dystrophy/neurodegenerative disorder clinics).
Diagnostic evaluation
Each patient will undergo a diagnostic evaluation tailored to their own particular presenting signs and symptoms. The geneticist will establish a differential diagnosis and recommend appropriate testing. These tests might evaluate for chromosomal disorders, inborn errors of metabolism, or single gene disorders.[citation needed]
Chromosome studies are used in the general genetics clinic to determine a cause for developmental delay or intellectual disability, birth defects, dysmorphic features, or autism.[citation needed] Chromosome analysis is also performed in the prenatal setting to determine whether a fetus is affected with aneuploidy or other chromosome rearrangements. Finally, chromosome abnormalities are often detected in cancer samples. A large number of different methods have been developed for chromosome analysis:
Chromosome analysis using a karyotype involves special stains that generate light and dark bands, allowing identification of each chromosome under a microscope.
Fluorescence in situ hybridization (FISH) involves fluorescent labeling of probes that bind to specific DNA sequences, used for identifying aneuploidy, genomic deletions or duplications, characterizing chromosomal translocations and determining the origin of ring chromosomes.
Chromosome painting is a technique that uses fluorescent probes specific for each chromosome to differentially label each chromosome. This technique is more often used in cancer cytogenetics, where complex chromosome rearrangements can occur.
Array comparative genomic hybridization is a newer molecular technique that involves hybridization of an individual DNA sample to a glass slide or microarray chip containing molecular probes (ranging from large ~200kb bacterial artificial chromosomes to small oligonucleotides) that represent unique regions of the genome. This method is particularly sensitive for detection of genomic gains or losses across the genome but does not detect balanced translocations or distinguish the location of duplicated genetic material (for example, a tandem duplication versus an insertional duplication).
Basic metabolic studies
Biochemical studies are performed to screen for imbalances of metabolites in the bodily fluid, usually the blood (plasma/serum) or urine, but also in cerebrospinal fluid (CSF). Specific tests of enzyme function (either in leukocytes, skin fibroblasts, liver, or muscle) are also employed under certain circumstances. In the US, the newborn screen incorporates biochemical tests to screen for treatable conditions such as galactosemia and phenylketonuria (PKU). Patients suspected to have a metabolic condition might undergo the following tests:[citation needed]
Urine organic acid analysis can be either performed using quantitative or qualitative methods, but in either case the test is used to detect the excretion of abnormal organic acids. These compounds are normally produced during bodily metabolism of amino acids and odd-chain fatty acids, but accumulate in patients with certain metabolic conditions.
The acylcarnitine combination profile detects compounds such as organic acids and fatty acids conjugated to carnitine. The test is used for detection of disorders involving fatty acid metabolism, including MCAD.
Pyruvate and lactate are byproducts of normal metabolism, particularly during anaerobic metabolism. These compounds normally accumulate during exercise or ischemia, but are also elevated in patients with disorders of pyruvate metabolism or mitochondrial disorders.
Ammonia is an end product of amino acid metabolism and is converted in the liver to urea through a series of enzymatic reactions termed the urea cycle. Elevated ammonia can therefore be detected in patients with urea cycle disorders, as well as other conditions involving liver failure.
Enzyme testing is performed for a wide range of metabolic disorders to confirm a diagnosis suspected based on screening tests.
Molecular studies
DNA sequencing is used to directly analyze the genomic DNA sequence of a particular gene. In general, only the parts of the gene that code for the expressed protein (exons) and small amounts of the flanking untranslated regions and introns are analyzed. Therefore, although these tests are highly specific and sensitive, they do not routinely identify all of the mutations that could cause disease.[citation needed]
Each cell of the body contains the hereditary information (DNA) wrapped up in structures called chromosomes. Since genetic syndromes are typically the result of alterations of the chromosomes or genes, there is no treatment currently available that can correct the genetic alterations in every cell of the body. Therefore, there is currently no "cure" for genetic disorders. However, for many genetic syndromes there is treatment available to manage the symptoms. In some cases, particularly inborn errors of metabolism, the mechanism of disease is well understood and offers the potential for dietary and medical management to prevent or reduce the long-term complications. In other cases, infusion therapy is used to replace the missing enzyme. Current research is actively seeking to use gene therapy or other new medications to treat specific genetic disorders.[citation needed]
Management of metabolic disorders
In general, metabolic disorders arise from enzyme deficiencies that disrupt normal metabolic pathways. For instance, in the hypothetical example:
A ⟶ B ⟶ C ⟶ D AAAA ⟶ BBBBBB ⟶ CCCCCCCCCC ⟶ (no D) X Y Z X Y | (no or insufficient Z) EEEEE
Compound "A" is metabolized to "B" by enzyme "X", compound "B" is metabolized to "C" by enzyme "Y", and compound "C" is metabolized to "D" by enzyme "Z". If enzyme "Z" is missing, compound "D" will be missing, while compounds "A", "B", and "C" will build up. The pathogenesis of this particular condition could result from lack of compound "D", if it is critical for some cellular function, or from toxicity due to excess "A", "B", and/or "C", or from toxicity due to the excess of "E" which is normally only present in small amounts and only accumulates when "C" is in excess. Treatment of the metabolic disorder could be achieved through dietary supplementation of compound "D" and dietary restriction of compounds "A", "B", and/or "C" or by treatment with a medication that promoted disposal of excess "A", "B", "C" or "E". Another approach that can be taken is enzyme replacement therapy, in which a patient is given an infusion of the missing enzyme "Z" or cofactor therapy to increase the efficacy of any residual "Z" activity.[citation needed]
Diet
Dietary restriction and supplementation are key measures taken in several well-known metabolic disorders, including galactosemia, phenylketonuria (PKU), maple syrup urine disease, organic acidurias and urea cycle disorders. Such restrictive diets can be difficult for the patient and family to maintain, and require close consultation with a nutritionist who has special experience in metabolic disorders. The composition of the diet will change depending on the caloric needs of the growing child and special attention is needed during a pregnancy if a woman is affected with one of these disorders.[citation needed]
Medication
Medical approaches include enhancement of residual enzyme activity (in cases where the enzyme is made but is not functioning properly), inhibition of other enzymes in the biochemical pathway to prevent buildup of a toxic compound, or diversion of a toxic compound to another form that can be excreted. Examples include the use of high doses of pyridoxine (vitamin B6) in some patients with homocystinuria to boost the activity of the residual cystathione synthase enzyme, administration of biotin to restore activity of several enzymes affected by deficiency of biotinidase, treatment with NTBC in Tyrosinemia to inhibit the production of succinylacetone which causes liver toxicity, and the use of sodium benzoate to decrease ammonia build-up in urea cycle disorders.[citation needed]
Certain lysosomal storage diseases are treated with infusions of a recombinant enzyme (produced in a laboratory), which can reduce the accumulation of the compounds in various tissues. Examples include Gaucher disease, Fabry disease, Mucopolysaccharidoses and Glycogen storage disease type II. Such treatments are limited by the ability of the enzyme to reach the affected areas (the blood brain barrier prevents enzyme from reaching the brain, for example), and can sometimes be associated with allergic reactions. The long-term clinical effectiveness of enzyme replacement therapies vary widely among different disorders.
Other examples
Angiotensin receptor blockers in Marfan syndrome & Loeys-Dietz
There are a variety of career paths within the field of medical genetics, and naturally the training required for each area differs considerably. The information included in this section applies to the typical pathways in the United States and there may be differences in other countries. US practitioners in clinical, counseling, or diagnostic subspecialties generally obtain board certification through the American Board of Medical Genetics.
A clinical geneticist is typically a physician who evaluates patients in the office or as a hospital consultation. This process includes a medical history, family history (pedigree), a detailed physical examination, reviewing objective data such as imaging and test results, establishing a differential diagnosis, and recommending appropriate diagnostic tests.
College (4 yrs) → Medical school (4 yrs) → Primary residency (1 yr) → Residency in Clinical genetics (2 yrs). Some Clinical geneticists also obtain a PhD degree (4-7 yrs). A new residency track offers a 4-year primary residency in Clinical genetics immediately after finishing Medical school.[citation needed]
Genetic counselor
MS
A genetic counselor specializes in the communication of genetic information to patients and families. Genetic counselors often work closely with Clinical geneticists or other physicians (such as Obstetricians or Oncologists) and often convey the results of the recommended tests.
College (4 yrs) → Graduate program in Genetic counseling (2 yrs).
Metabolic nurse and/or nutritionist
BA/BS, MS, RN
One of the critical aspects of the management of patients with metabolic disorders is the appropriate nutritional intervention (either restricting the compound that cannot be metabolized or supplementing deficient compounds as the result of an enzyme deficiency). The metabolic nurse and nutritionist play important roles in coordinating dietary management.
College (4 yrs) → Nursing school or graduate training in nutrition.
Biochemical diagnostics
BS, MS, PhD, MBBS, MD, DO, DO-PhD, or MD-PhD
Individuals who specialize in Biochemical genetics typically work in the diagnostic laboratory, analyzing and interpreting specialized biochemical tests that measure amino acids, organic acids, and enzyme activity. Some Clinical Geneticists are also board-certified in Biochemical Genetics.
College (4 yrs) → Graduate school (PhD, usually 4–7 years) and/or Medical school (4 years)
Cytogenetic diagnostics
BS, MS, PhD, MBBS, MD, DO, DO-PhD, or MD-PhD
Individuals who specialize in Cytogenetics typically work in the diagnostic laboratory, analyzing and interpreting karyotypes, FISH, and comparative genomic hybridization tests. Some Clinical Geneticists are also board-certified in Cytogenetics.
College (4 yrs) → Graduate school (PhD, usually 4–7 years) and/or Medical school (4 years)
Molecular genetics
BS, MS, PhD, MBBS, MD, DO, DO-PhD, or MD-PhD
Individuals who specialize in Molecular genetics typically work in the diagnostic laboratory, analyzing and interpreting specialized genetic tests that look for disease-causing changes (mutations) in the DNA. Some examples of molecular diagnostic tests include DNA sequencing and Southern blotting.
College (4 yrs) → Graduate school (PhD, usually 4–7 years) and/or Medical school (4 years)
Research geneticist
BS, MS, PhD, MBBS, MD, DO, DO-PhD, or MD-PhD
Any researcher who studies the genetic basis of human disease or uses model organisms to study disease mechanisms could be considered a Research Geneticist. Many of the clinical career paths also include basic or translational research, and thus individuals in the field of medical genetics often participate in some form of research.
College (4 yrs) → Graduate school (PhD, usually 4–7 years) and/or Medical school (4 years) → Post-doctoral research training (usually 3+ years)
Laboratory technician
AS, BS, MS
Technicians in the diagnostic or research labs handle samples and run the assays at the bench.
College (4 yrs), may have higher degree (MS, 2+ years)
Ethical, legal and social implications
Genetic information provides a unique type of knowledge about an individual and his/her family, fundamentally different from a typically laboratory test that provides a "snapshot" of an individual's health status. The unique status of genetic information and inherited disease has a number of ramifications with regard to ethical, legal, and societal concerns.
On 19 March 2015, scientists urged a worldwide ban on clinical use of methods, particularly the use of CRISPR and zinc finger, to edit the human genome in a way that can be inherited.[7][8][9][10] In April 2015 and April 2016, Chinese researchers reported results of basic research to edit the DNA of non-viable human embryos using CRISPR.[11][12][13] In February 2016, British scientists were given permission by regulators to genetically modify human embryos by using CRISPR and related techniques on condition that the embryos were destroyed within seven days.[14] In June 2016 the Dutch government was reported to be planning to follow suit with similar regulations which would specify a 14-day limit.[15]
Medical genetics is recognized as a distinct medical specialty. In the U.S., medical genetics has its own approved board (the American Board of Medical Genetics) and clinical specialty college (the American College of Medical Genetics). The college holds an annual scientific meeting, publishes a monthly journal, Genetics in Medicine, and issues position papers and clinical practice guidelines on a variety of topics relevant to human genetics.[citation needed]
The broad range of research in medical genetics reflects the overall scope of this field, including basic research on genetic inheritance and the human genome, mechanisms of genetic and metabolic disorders, translational research on new treatment modalities, and the impact of genetic testing
Basic genetics research
Basic research geneticists usually undertake research in universities, biotechnology firms and research institutes.
Sometimes the link between a disease and an unusual gene variant is more subtle. The genetic architecture of common diseases is an important factor in determining the extent to which patterns of genetic variation influence group differences in health outcomes.[16][17][18] According to the common disease/common variant hypothesis, common variants present in the ancestral population before the dispersal of modern humans from Africa play an important role in human diseases.[19] Genetic variants associated with Alzheimer disease, deep venous thrombosis, Crohn disease, and type 2 diabetes appear to adhere to this model.[20] However, the generality of the model has not yet been established and, in some cases, is in doubt.[17][21][22] Some diseases, such as many common cancers, appear not to be well described by the common disease/common variant model.[23]
Another possibility is that common diseases arise in part through the action of combinations of variants that are individually rare.[24][25] Most of the disease-associated alleles discovered to date have been rare, and rare variants are more likely than common variants to be differentially distributed among groups distinguished by ancestry.[23][26] However, groups could harbor different, though perhaps overlapping, sets of rare variants, which would reduce contrasts between groups in the incidence of the disease.
The number of variants contributing to a disease and the interactions among those variants also could influence the distribution of diseases among groups. The difficulty that has been encountered in finding contributory alleles for complex diseases and in replicating positive associations suggests that many complex diseases involve numerous variants rather than a moderate number of alleles, and the influence of any given variant may depend in critical ways on the genetic and environmental background.[21][27][28][29] If many alleles are required to increase susceptibility to a disease, the odds are low that the necessary combination of alleles would become concentrated in a particular group purely through drift.[30]
One area in which population categories can be important considerations in genetics research is in controlling for confounding between population substructure, environmental exposures, and health outcomes. Association studies can produce spurious results if cases and controls have differing allele frequencies for genes that are not related to the disease being studied,[31] although the magnitude of this problem in genetic association studies is subject to debate.[32][33] Various methods have been developed to detect and account for population substructure,[34][35] but these methods can be difficult to apply in practice.[36]
Population substructure also can be used to advantage in genetic association studies.[37] For example, populations that represent recent mixtures of geographically separated ancestral groups can exhibit longer-range linkage disequilibrium between susceptibility alleles and genetic markers than is the case for other populations.[38][39][40][41] Genetic studies can use this admixture linkage disequilibrium to search for disease alleles with fewer markers than would be needed otherwise. Association studies also can take advantage of the contrasting experiences of racial or ethnic groups, including migrant groups, to search for interactions between particular alleles and environmental factors that might influence health.[42][43]
↑Rose, Nikolas. (2009). The Politics of Life Itself: Biomedicine, Power, and Subjectivity in the Twenty-First Century. Princeton University Press. ISBN978-0-691-12190-1. OCLC995257497.
↑Lohmueller KE, Pearce CL, Pike M, Lander ES, Hirschhorn JN (2003). "Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease". Nat Genet. 33 (2): 177–182. doi:10.1038/ng1071. PMID12524541. S2CID6850292.
12Weiss KM, Terwilliger JD (2000). "How many diseases does it take to map a gene with SNPs?". Nat Genet. 26 (2): 151–157. doi:10.1038/79866. PMID11017069. S2CID685795.
↑Risch N, Burchard E, Ziv E, Tang H, "Categorization of humans in biomedical research: genes, race and disease", Genome Biol (2002) 3 (http://genomebiology.com/2002/3/7/comment/2007Archived 2006-06-24 at the Wayback Machine ) (electronically published July 1, 2002; accessed August 25, 2005)
↑Cooper RS, "Genetic factors in ethnic disparities in health", in Anderson NB, Bulatao RA, Cohen B, eds., Critical perspectives on racial and ethnic differences in health in later life, (Washington DC: National Academy Press, 2004), 267–309.
↑Thomas DC, Witte JS (2002). "Point: population stratification: a problem for case-control studies of candidate-gene associations?". Cancer Epidemiol Biomarkers Prev. 11 (6): 505–512. PMID12050090.
↑Wacholder S, Rothman N, Caporaso N (2002). "Counterpoint: bias from population stratification is not a major threat to the validity of conclusions from epidemiological studies of common polymorphisms and cancer". Cancer Epidemiol Biomarkers Prev. 11 (6): 513–520. PMID12050091.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.