
Lesch–Nyhan syndrome (LNS) is a rare inherited disorder caused by a deficiency of the enzyme hypoxanthine-guanine phosphoribosyltransferase (HGPRT). This deficiency occurs due to mutations in the HPRT1 gene located on the X chromosome. LNS affects about 1 in 380,000 live births. The disorder was first recognized and clinically characterized by American medical student Michael Lesch and his mentor, pediatrician William Nyhan, at Johns Hopkins.
In genetics, anticipation is a phenomenon whereby as a genetic disorder is passed on to the next generation, the symptoms of the genetic disorder become apparent at an earlier age with each generation. In most cases, an increase in the severity of symptoms is also noted. Anticipation is common in trinucleotide repeat disorders, such as Huntington's disease and myotonic dystrophy, where a dynamic mutation in DNA occurs. All of these diseases have neurological symptoms. Prior to the understanding of the genetic mechanism for anticipation, it was debated whether anticipation was a true biological phenomenon or whether the earlier age of diagnosis was related to heightened awareness of disease symptoms within a family.
Macroorchidism is a disorder found in males, specifically in children, where a subject has abnormally large testes. The condition is commonly inherited in connection with fragile X syndrome (FXS), which is also the second most common genetic cause of intellectual disability. The condition is also a rare sign of McCune–Albright syndrome. The opposite of macroorchidism is called microorchidism, which is the condition of abnormally small testes.
In genetics, trinucleotide repeat disorders, a subset of microsatellite expansion diseases, are a set of over 30 genetic disorders caused by trinucleotide repeat expansion, a kind of mutation in which repeats of three nucleotides increase in copy numbers until they cross a threshold above which they cause developmental, neurological or neuromuscular disorders. In addition to the expansions of these trinucleotide repeats, expansions of one tetranucleotide (CCTG), five pentanucleotide, three hexanucleotide, and one dodecanucleotide (CCCCGCCCCGCG) repeat cause 13 other diseases. Depending on its location, the unstable trinucleotide repeat may cause defects in a protein encoded by a gene; change the regulation of gene expression; produce a toxic RNA, or lead to production of a toxic protein. In general, the larger the expansion the faster the onset of disease, and the more severe the disease becomes.
The Sherman paradox was a term used to describe the anomalous pattern of inheritance found in fragile X syndrome. The phenomenon is also referred to as anticipation or dynamic mutation.

Autism has multiple causes. This article focuses on heritable causes. The heritability of autism is the proportion of differences in expression of autism that can be explained by genetic variation; if the heritability of a condition is high, then the condition is considered to be primarily genetic. Autism has a strong genetic basis. Although the genetics of autism are complex, autism spectrum disorder (ASD) is explained more by multigene effects than by rare mutations with large effects.

FMR1 is a human gene that codes for a protein called fragile X messenger ribonucleoprotein, or FMRP. This protein, most commonly found in the brain, is essential for normal cognitive development and female reproductive function. Mutations of this gene can lead to fragile X syndrome, intellectual disability, premature ovarian failure, autism, Parkinson's disease, developmental delays and other cognitive deficits. The FMR1 premutation is associated with a wide spectrum of clinical phenotypes that affect more than two million people worldwide.
A trinucleotide repeat expansion, also known as a triplet repeat expansion, is the DNA mutation responsible for causing any type of disorder categorized as a trinucleotide repeat disorder. These are labelled in dynamical genetics as dynamic mutations. Triplet expansion is caused by slippage during DNA replication, also known as "copy choice" DNA replication. Due to the repetitive nature of the DNA sequence in these regions, 'loop out' structures may form during DNA replication while maintaining complementary base pairing between the parent strand and daughter strand being synthesized. If the loop out structure is formed from the sequence on the daughter strand this will result in an increase in the number of repeats. However, if the loop out structure is formed on the parent strand, a decrease in the number of repeats occurs. It appears that expansion of these repeats is more common than reduction. Generally, the larger the expansion the more likely they are to cause disease or increase the severity of disease. Other proposed mechanisms for expansion and reduction involve the interaction of RNA and DNA molecules.

Fragile X-associated tremor/ataxia syndrome (FXTAS) is a late-onset neurodegenerative disorder most frequently seen in male premutation carriers of Fragile X syndrome (FXS) over the age of 50. The main clinical features of FXTAS include problems of movement with cerebellar gait ataxia and action tremor. Associated features include parkinsonism, cognitive decline, and dysfunction of the autonomic nervous system. FXTAS is found in Fragile X "premutation" carriers, which is defined as a trinucleotide repeat expansion of 55-200 CGG repeats in the Fragile X mental retardation-1 (FMR1) gene. 4-40 CGG repeats in this gene is considered normal, while individual with >200 repeats have full Fragile X Syndrome.

Sotos syndrome is a rare genetic disorder characterized by excessive physical growth during the first years of life. Excessive growth often starts in infancy and continues into the early teen years. The disorder may be accompanied by autism, mild intellectual disability, delayed motor, cognitive, and social development, hypotonia, and speech impairments. Children with Sotos syndrome tend to be large at birth and are often taller, heavier, and have relatively large skulls (macrocephaly) than is normal for their age. Signs of the disorder, which vary among individuals, include a disproportionately large skull with a slightly protrusive forehead, large hands and feet, large mandible, hypertelorism, and downslanting eyes. Clumsiness, an awkward gait, and unusual aggressiveness or irritability may also occur.

In molecular biology, FMR1 antisense RNA 1 (FMR1-AS1), also known as ASFMR1 or FMR4, is a long non-coding RNA. The FMR1-AS1 gene overlaps, and is antisense to, the CGG repeat region of the FMR1 gene. Its expression is upregulated in fragile X syndrome premutation carriers, and silenced in patients with fragile X syndrome. FMR1-AS1 has an anti-apoptotic function.
Autism spectrum disorder (ASD) refers to a variety of conditions typically identified by challenges with social skills, communication, speech, and repetitive sensory-motor behaviors. The 11th International Classification of Diseases (ICD-11), released in January 2021, characterizes ASD by the associated deficits in the ability to initiate and sustain two-way social communication and restricted or repetitive behavior unusual for the individual's age or situation. Although linked with early childhood, the symptoms can appear later as well. Symptoms can be detected before the age of two and experienced practitioners can give a reliable diagnosis by that age. However, official diagnosis may not occur until much older, even well into adulthood. There is a large degree of variation in how much support a person with ASD needs in day-to-day life. This can be classified by a further diagnosis of ASD level 1, level 2, or level 3. Of these, ASD level 3 describes people requiring very substantial support and who experience more severe symptoms. ASD-related deficits in nonverbal and verbal social skills can result in impediments in personal, family, social, educational, and occupational situations. This disorder tends to have a strong correlation with genetics along with other factors. More research is identifying ways in which epigenetics is linked to autism. Epigenetics generally refers to the ways in which chromatin structure is altered to affect gene expression. Mechanisms such as cytosine regulation and post-translational modifications of histones. Of the 215 genes contributing, to some extent in ASD, 42 have been found to be involved in epigenetic modification of gene expression. Some examples of ASD signs are specific or repeated behaviors, enhanced sensitivity to materials, being upset by changes in routine, appearing to show reduced interest in others, avoiding eye contact and limitations in social situations, as well as verbal communication. When social interaction becomes more important, some whose condition might have been overlooked suffer social and other exclusion and are more likely to have coexisting mental and physical conditions. Long-term problems include difficulties in daily living such as managing schedules, hypersensitivities, initiating and sustaining relationships, and maintaining jobs.
Fragile X-associated primary ovarian insufficiency (FXPOI) is the most common genetic cause of premature ovarian failure in women with a normal karyotype 46,XX. The expansion of a CGG repeat in the 5' untranslated region of the FMR1 gene from the normal range of 5-45 repeats to the premutation range of 55-199 CGGs leads to risk of FXPOI for ovary-bearing individuals. About 1:150-1:200 women in the US population carry a premutation. Women who carry an FMR1 premutation have a roughly 20% risk of being diagnosed with FXPOI, compared to 1% for the general population, and an 8-15% risk of developing the neurogenerative tremor/ataxia disorder (FXTAS). FMR1 premutation women are also at increased risk of having a child with a CGG repeat that is expanded to >200 repeats. Individuals with a full mutation, unlike the premutation, produce little to no mRNA or protein from the FMR1 gene and have fragile X syndrome as a result.

Metadoxine, also known as pyridoxine-pyrrolidone carboxylate, is a drug used to treat chronic and acute alcohol intoxication. Metadoxine accelerates alcohol clearance from the blood.
Nagwa Abdel Meguid is an Egyptian geneticist and 2002 winner of the L’Oreal UNESCO Award for Women in Science for Africa and the Middle East. Her research has "identified several genetic mutations that cause common syndromes such as the fragile X syndrome and Autism".

MECP2 duplication syndrome (M2DS) is a rare disease that is characterized by severe intellectual disability and impaired motor function. It is an X-linked genetic disorder caused by the overexpression of MeCP2 protein.
ADNP syndrome, also known as Helsmoortel-Van der Aa syndrome (HVDAS), is a non-inherited neurodevelopmental disorder caused by mutations in the activity-dependent neuroprotector homeobox (ADNP) gene.
David L. Nelson is an American human geneticist, currently an associate director at the Intellectual and Developmental Disabilities Research Center (1995), and professor at the Department of Molecular and Human Genetics at Baylor College of Medicine BCM since 1999. Since 2018, he is the director at the Cancer and Cell Biology Ph.D program, and the director of Integrative Molecular and Biomedical Sciences Ph.D since 2015 at BCM.
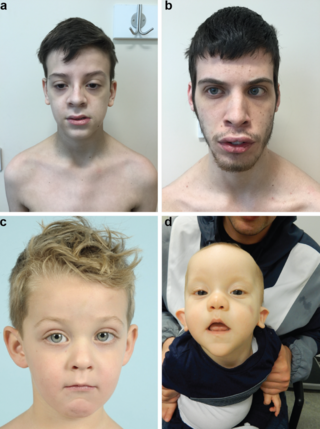
Beck–Fahrner syndrome, also known as BEFAHRS and TET3 deficiency, is a rare genetic disorder caused by mutations of the TET3 gene. The clinical presentation varies among individuals, but typically includes global developmental delay, slow progress in mental and physical activities, autism, decreased muscle tone, epilepsy and dysmorphic features.
Unstable DNA sequence are segments of genetic material that exhibit high rates of mutation or variation over time, resulting in significant genetic diversity within populations or even individual organisms.


