
Anemia or anaemia is a blood disorder in which the blood has a reduced ability to carry oxygen. This can be due to a lower than normal number of red blood cells, a reduction in the amount of hemoglobin available for oxygen transport, or abnormalities in hemoglobin that impair its function.

A myelodysplastic syndrome (MDS) is one of a group of cancers in which immature blood cells in the bone marrow do not mature, and as a result, do not develop into healthy blood cells. Early on, no symptoms typically are seen. Later, symptoms may include fatigue, shortness of breath, bleeding disorders, anemia, or frequent infections. Some types may develop into acute myeloid leukemia.

Fanconi anemia (FA) is a rare, autosomal recessive, genetic disease resulting in impaired response to DNA damage in the FA/BRCA pathway. Although it is a very rare disorder, study of this and other bone marrow failure syndromes has improved scientific understanding of the mechanisms of normal bone marrow function and development of cancer. Among those affected, the majority develop cancer, most often acute myelogenous leukemia (AML), MDS, and liver tumors. 90% develop aplastic anemia by age 40. About 60–75% have congenital defects, commonly short stature, abnormalities of the skin, arms, head, eyes, kidneys, and ears, and developmental disabilities. Around 75% have some form of endocrine problem, with varying degrees of severity. 60% of FA is FANC-A, 16q24.3, which has later onset bone marrow failure.

Hereditary spherocytosis (HS) is a congenital hemolytic disorder wherein a genetic mutation coding for a structural membrane protein phenotype causes the red blood cells to be sphere-shaped (spherocytosis), rather than the normal biconcave disk shape. This abnormal shape interferes with the cells' ability to flex during blood circulation, and also makes them more prone to rupture under osmotic stress, mechanical stress, or both. Cells with the dysfunctional proteins are degraded in the spleen, which leads to a shortage of erythrocytes and results in hemolytic anemia.

Erythropoiesis is the process which produces red blood cells (erythrocytes), which is the development from erythropoietic stem cell to mature red blood cell.
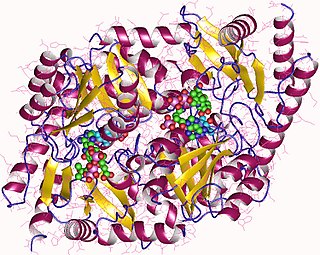
Aminolevulinic acid synthase (ALA synthase, ALAS, or delta-aminolevulinic acid synthase) is an enzyme (EC 2.3.1.37) that catalyzes the synthesis of δ-aminolevulinic acid (ALA) the first common precursor in the biosynthesis of all tetrapyrroles such as hemes, cobalamins and chlorophylls. The reaction is as follows:

Chromosome 5q deletion syndrome is an acquired, hematological disorder characterized by loss of part of the long arm of human chromosome 5 in bone marrow myelocyte cells. This chromosome abnormality is most commonly associated with the myelodysplastic syndrome.
Pancytopenia is a medical condition in which there is significant reduction in the number of almost all blood cells.
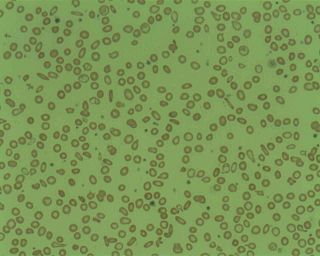
Microcytic anaemia is any of several types of anemia characterized by smaller than normal red blood cells. The normal mean corpuscular volume is approximately 80–100 fL. When the MCV is <80 fL, the red cells are described as microcytic and when >100 fL, macrocytic. The MCV is the average red blood cell size.
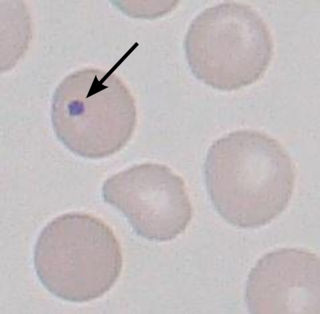
A Howell–Jolly body is a cytopathological finding of basophilic nuclear remnants in circulating erythrocytes. During maturation in the bone marrow, late erythroblasts normally expel their nuclei; but, in some cases, a small portion of DNA remains. The presence of Howell–Jolly bodies usually signifies a damaged or absent spleen, because a healthy spleen would normally filter such erythrocytes.

Macrocytosis is a condition where red blood cells are larger than normal. These enlarged cells, also known as macrocytes, are defined by a mean corpuscular volume (MCV) that exceeds the upper reference range established by the laboratory and hematology analyzer. Upon examination of a peripheral blood smear under microscope, these macrocytes appear larger than standard erythrocytes. It’s noteworthy that macrocytosis is a common morphological feature in neonatal peripheral blood. The presence of macrocytosis can indicate a range of conditions, from benign, treatable illnesses to more serious underlying disorders.

GATA-binding factor 1 or GATA-1 is the founding member of the GATA family of transcription factors. This protein is widely expressed throughout vertebrate species. In humans and mice, it is encoded by the GATA1 and Gata1 genes, respectively. These genes are located on the X chromosome in both species.

A promyelocyte is a granulocyte precursor, developing from the myeloblast and developing into the myelocyte. Promyelocytes measure 12–20 microns in diameter. The nucleus of a promyelocyte is approximately the same size as a myeloblast but their cytoplasm is much more abundant. They also have less prominent nucleoli than myeloblasts and their chromatin is more coarse and clumped. The cytoplasm is basophilic and contains primary red/purple granules.

Chronic myelomonocytic leukemia (CMML) is a type of leukemia, which are cancers of the blood-forming cells of the bone marrow. In adults, blood cells are formed in the bone marrow, by a process that is known as haematopoiesis. In CMML, there are increased numbers of monocytes and immature blood cells (blasts) in the peripheral blood and bone marrow, as well as abnormal looking cells (dysplasia) in at least one type of blood cell.
Copper deficiency, or hypocupremia, is defined either as insufficient copper to meet the needs of the body, or as a serum copper level below the normal range. Symptoms may include fatigue, decreased red blood cells, early greying of the hair, and neurological problems presenting as numbness, tingling, muscle weakness, and ataxia. The neurodegenerative syndrome of copper deficiency has been recognized for some time in ruminant animals, in which it is commonly known as "swayback". Copper deficiency can manifest in parallel with vitamin B12 and other nutritional deficiencies.

Pappenheimer bodies are abnormal basophilic granules of iron found inside red blood cells on routine blood stain. They are a type of inclusion body composed of ferritin aggregates, or mitochondria or phagosomes containing aggregated ferritin. They appear as dense, blue-purple granules within the red blood cell and there are usually only one or two, located in the cell periphery. They stain on a Romanowsky stain because clumps of ribosomes are co‐precipitated with the iron‐containing organelles.

Delta-aminolevulinate synthase 2 also known as ALAS2 is a protein that in humans is encoded by the ALAS2 gene. ALAS2 is an aminolevulinic acid synthase.
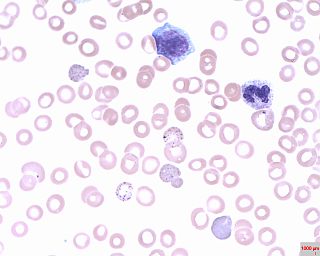
Basophilic stippling, also known as punctate basophilia, is the presence of numerous basophilic granules that are dispersed through the cytoplasm of erythrocytes in a peripheral blood smear. They can be demonstrated to be RNA. They are composed of aggregates of ribosomes; degenerating mitochondria and siderosomes may be included in the aggregates. In contrast to Pappenheimer bodies, they are negative with Perls' acid ferrocyanide stain for iron. Basophilic stippling is indicative of disturbed erythropoiesis. It can also be found in some normal individuals.

Glutaredoxin 5, also known as GLRX5, is a protein which in humans is encoded by the GLRX5 gene located on chromosome 14. This gene encodes a mitochondrial protein, which is evolutionarily conserved. It is involved in the biogenesis of iron- sulfur clusters, which are required for normal iron homeostasis. Mutations in this gene are associated with autosomal recessive pyridoxine-refractory sideroblastic anemia.
Bone marrow failure occurs in individuals who produce an insufficient amount of red blood cells, white blood cells or platelets. Red blood cells transport oxygen to be distributed throughout the body's tissue. White blood cells fight off infections that enter the body. Bone marrow progenitor cells known as megakaryocytes produce platelets, which trigger clotting, and thus help stop the blood flow when a wound occurs.

