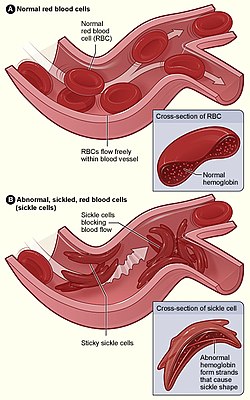
Figure (A) shows normal red blood cells flowing freely through a blood vessel. The inset shows a cross-section of a normal red blood cell with normal haemoglobin. Figure (B) shows abnormal, sickled red blood cells sticking at the branching point in a blood vessel. The inset image shows a cross-section of a sickle cell with long polymerised sickle haemoglobin (HbS) strands stretching and distorting the cell shape to look like a crescent moon.
34,000 p.a. (a contributory factor to a further 376,000 p.a.)[9]
Sickle cell disease (SCD), also simply called sickle cell, is a group of inherited haemoglobin-relatedblood disorders.[2] The most common type is known as sickle cell anaemia.[2] Sickle cell anaemia results in an abnormality in the oxygen-carrying protein haemoglobin found in red blood cells.[2] This leads to the red blood cells adopting an abnormal sickle-like shape under certain circumstances. With this shape, they are unable to deform as they pass through capillaries, causing blockages.[2]
Problems in sickle cell disease typically begin around 5 to 6 months of age.[1] Several health problems may develop, such as attacks of pain (known as a sickle cell crisis) in joints, anaemia, swelling in the hands and feet, bacterial infections, dizziness[10] and stroke.[1] The probability of severe symptoms, including long-term pain, increases with age.[2] Without treatment, people with sickle cell disease rarely reach adulthood, but with good healthcare, median life expectancy is between 58 and 66 years.[11][12] All of the major organs are affected by sickle cell disease. The liver, heart, kidneys, lungs, gallbladder, eyes, bones, and joints can be damaged from the abnormal functions of the sickle cells and their inability to effectively flow through the small blood vessels.[13]
Sickle cell disease occurs when a person inherits two abnormal copies of the β-globin gene that make haemoglobin, one from each parent.[14] The abnormal gene generates haemoglobin S (HbS) which changes the properties of red blood cells.[2] A sickle cell crisis occurs when red blood cells switch from the normal saucer-like shape to a sickle-like shape which can obstruct small blood vessels; an attack can be set off by temperature changes, stress, dehydration, and high altitude.[1] A person with a single abnormal gene does not usually have symptoms and is said to have sickle cell trait,[14] these people are also referred to as carriers.[7] Diagnosis is by a blood test, and some countries test all babies at birth for the disease.[6] Diagnosis of the unborn foetus is also possible during pregnancy.[6]
As of 2021[update], sickle cell disease is estimated to affect about 7.7 million people worldwide, directly causing an estimated 34,000 annual deaths and a contributory factor to a further 376,000 deaths.[9][18] About 80% of sickle cell disease cases are believed to occur in sub-Saharan Africa.[19] It also occurs to a lesser degree among people in parts of India, Southern Europe, West Asia, North Africa and among people of African origin (sub-Saharan) living in other parts of the world.[20] The condition was first described in the medical literature by American physician James B. Herrick in 1910.[21][22] In 1949, its genetic transmission was determined by E. A. Beet and J. V. Neel.[22] In 1954, it was established that carriers of the abnormal gene are protected to some degree against malaria,[22] which accounts for its persistence in populations threatened by malaria.[23]
Signs and symptoms
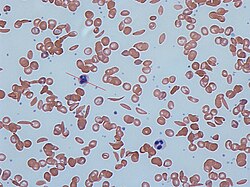
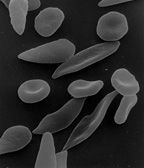
Dactylitis in the hands of an infantSickle cells in human blood - both normal red blood cells and sickle-shaped cells are present.Normal blood cells next to a sickle blood cell, coloured scanning electron microscope image
Signs of sickle cell disease usually begin in early childhood. The severity of symptoms can vary from person to person, as can the frequency of crisis events.[24][19] Sickle cell disease may lead to various acute and chronic complications, several of which have a high mortality rate.[25]
First events
When sickle cell disease presents within the first year of life, the most common problem is an episode of pain and swelling in the child's hands and feet, known as dactylitis or "hand-foot syndrome". Pallor, jaundice, and fatigue can also be early signs due to anaemia resulting from sickle cell disease.[26]
In children older than 2 years, the most common initial presentation is a painful episode of a generalised or variable nature, while a slightly less common presentation involves acute chest pain. Dactylitis is rare or almost never occurs in children over the age of 2.[26][1]
Critical events
Vaso-occlusive crisis
Also termed "sickle cell crisis" or "sickling crisis", the vaso-occlusive crisis (VOC) manifests principally as extreme pain, most often affecting the chest, back, legs, and/or arms.[27] The underlying cause is sickle-shaped red blood cells that obstruct capillaries and restrict blood flow to an organ, resulting in ischaemia, pain, necrosis, and often organ damage. The frequency, severity, and duration of these crises vary considerably. Milder crises can be managed with nonsteroidal anti-inflammatory drugs. For more severe crises, patients may require inpatient management for intravenous opioids. Vaso-occlusive crisis involving organs such as the lungs or the penis are considered an emergency and treated with red blood cell transfusions.[28]
A VOC can be triggered by anything which causes blood vessels to constrict; this includes physical or mental stress, cold, and dehydration.[29] "After Haemoglobin S (HbS) deoxygenates in the capillaries, it takes some time (seconds) for HbS polymerization and the subsequent flexible-to-rigid transformation. If the transit time of RBC through the microvasculature is longer than the polymerization time, sickled RBC will lodge in the microvasculature."[30]
Splenic sequestration crisis
The spleen is especially prone to damage in sickle cell disease due to its role as a blood filter. A splenic sequestration crisis, also known as a spleen crisis, is a medical emergency that occurs when sickled red blood cells block the spleen's filter mechanism, causing the spleen to swell and fill with blood. The accumulation of red blood cells in the spleen results in a sudden drop in circulating haemoglobin and potentially life-threatening anaemia. Symptoms include pain on the left side, swollen spleen (which can be detected by palpation), fatigue, dizziness, irritability, rapid heartbeat, or pale skin. It most commonly affects young children; the median age of first occurrence is 1.4 years. By the age of 5 years, repeated instances of sequestration cause scarring and eventual atrophy of the spleen.[31][32][33]
Treatment is supportive, with blood transfusion if haemoglobin levels fall too low. Full or partial splenectomy may be necessary.[34] Long term consequences of a loss of spleen function are increased susceptibility to bacterial infections.[33]
Acute chest syndrome
Acute chest syndrome is caused by a VOC which affects the lungs, possibly triggered by infection or by emboli which have circulated from other organs. Symptoms include wheezing, chest pain, fever, pulmonary infiltrate (visible on x-ray), and hypoxemia. After sickling crisis (see above), it is the second-most common cause of hospitalisation, and it accounts for about 25% of deaths in patients with sickle cell disease. Most cases present with vaso-occlusive crises and then develop acute chest syndrome.[35][36]
Aplastic crisis
Aplastic crises are instances of an acute worsening of the patient's baseline anaemia, producing pale appearance, fast heart rate, and fatigue. This crisis is normally triggered by parvovirus B19, which directly affects production of red blood cells by invading the red cell precursors and multiplying in and destroying them.[37] Parvovirus infection [38] almost completely prevents red blood cell production for two to three days (red cell aplasia). In normal individuals, this is of little consequence, but the shortened red cell life of people with sickle cell disease results in an abrupt, life-threatening situation. Reticulocyte count drops dramatically during the disease (causing reticulocytopenia), red cell production lapses, and the rapid destruction of existing red cells leads to acute and severe anaemia. This crisis takes four to seven days to resolve. Most patients can be managed supportively; some need a blood transfusion.[39]
Complications
Sickle cell anaemia can lead to various complications, including:[40]
An increased risk of severe bacterial infections is due to the loss of functioning spleen tissue. These infections are typically caused by bacteria such as Streptococcus pneumoniae and Haemophilus influenzae. Daily penicillin prophylaxis is the most commonly used treatment during childhood, with some haematologists continuing treatment indefinitely. Patients benefit from routine vaccination for S. pneumoniae.[41]
Stroke can result from blockage of blood vessels in the brain, causing numbness, confusion, or weakness, which may be long-lasting. Silent stroke causes no immediate symptoms, but is associated with damage to the brain. Silent stroke is probably five times as common as symptomatic stroke. About 10–15% of children with sickle cell disease have strokes, with silent strokes predominating in the younger patients.[40][42][43]
Chronic pain: Even in the absence of acute vaso-occlusive pain, many patients have unreported chronic pain.[51]
Pulmonary hypertension (increased pressure on the pulmonary artery) can lead to strain on the right ventricle and a risk of heart failure; typical symptoms are shortness of breath, decreased exercise tolerance, and episodes of syncope.[52] Evidence of pulmonary hypertension is found in 21% of children and 30% of adults when tested; this is associated with reduced walking distance and increased mortality.[53]
Autosomal recessive inheritance means acquiring two changed genes from each parent. If both parents are carriers for the autosomal recessive gene, there is a 25% chance of their child having and expressing the disorder. Other children will be unaffected, but may be carriers.Base-pair substitution that causes sickle cell anaemia
Haemoglobin is an oxygen-binding protein, found in erythrocytes, which transports oxygen from the lungs (or in the foetus, from the placenta) to the tissues. Each molecule of haemoglobin comprises 4 protein subunits, referred to as globins.[57] Normally, humans have:-
haemoglobin F (foetal haemoglobin, HbF), consisting of two alpha (α-globin) and two gamma (γ-globin) chains. This dominates during the development of the foetus and until about 6 weeks of age. Afterwards, haemoglobin A dominates throughout life.[58]
haemoglobin A (adult haemoglobin, HbA) which consists of two alpha and two beta (β-globin) chains. This is the most common human haemoglobin tetramer, accounting for over 97% of the total red blood cell haemoglobin in normal adults.[59]
Haemoglobin B2 (HbA2) is a second form of adult haemoglobin and is composed of two alpha and two delta (δ-globin) chains. This haemoglobin typically makes up 1–3% of haemoglobin in adults.[59]
β-globin is encoded by the HBB gene on human chromosome 11; mutations in this gene produce variants of the protein which are implicated with abnormal hemoglobins. The mutation that causes sickle cell disease results in an abnormal haemoglobin known as haemoglobin S (HbS), which replaces HbA in adults.[24] The human genome contains a pair of genes for β-globin; in people with sickle cell disease, both genes are affected, and the erythropoietic cells in the bone marrow will only create HbS. In people with sickle cell trait, only one gene is abnormal; erythropoiesis generates a mixture of normal HbA and sickle HbS. The person has very few, if any, symptoms of sickle cell disease but carries the gene and can pass it on to their children.[60]
Sickle cell disease has an autosomal recessive pattern of inheritance from parents.[61] Both copies of the affected gene must carry the same mutation (homozygous condition) for a person to be affected by an autosomal recessive disorder. An affected person usually has unaffected parents who each carry one mutated gene and one normal gene (heterozygous condition) and are referred to as genetic carriers; they may not have any symptoms.[62] When both parents have the sickle cell trait, any given child has a 25% chance of sickle cell disease; a 25% chance of no sickle cell alleles, and a 50% chance of the heterozygous condition (see diagram).[63]
There are several different haplotypes of the sickle cell gene mutation, indicating that it may have arisen spontaneously in different geographic areas[64]. The variants are known as Cameroon, Senegal, Benin, Bantu, and Saudi-Asian.[65] These are clinically important because some are associated with higher HbF levels, e.g., Senegal and Saudi-Asian variants, and tend to have milder disease.[66]
The gene defect is a single nucleotide mutation of the β-globin gene, which results in the amino acidglutamate being substituted by valine at position 6 of the β-globin chain.[67] Haemoglobin S with this mutation is referred to as HbS, as opposed to the normal adult HbA. Under conditions of normal oxygen concentration, this causes no apparent effects on the structure of haemoglobin or its ability to transport oxygen around the body. However, the deoxy form of HbS has an exposed hydrophobic patch, which causes HbS molecules to join to form long, inflexible chains. Under conditions of low oxygen concentration in the bloodstream, such as exercise, stress, altitude, or dehydration, HbS polymerisation forms fibrous precipitates within the red blood cell.[68] In people homozygous for the sickle cell mutation, the presence of long-chain polymers of HbS distort the shape of the red blood cell from a smooth, doughnut-like shape to the sickle shape, making it fragile and susceptible to blocking or breaking within capillaries.[67]
In people heterozygous for HbS (carriers of sickle cell disease), the polymerisation problems are minor because the normal allele can produce half of the haemoglobin. Sickle cell carriers have symptoms only if they are deprived of oxygen (for example, at altitude) or while severely dehydrated.[69]
Malaria
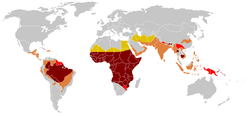
Historical distribution of the sickle cell traitHistorical distribution of malaria (no longer endemic in Europe)Modern distribution of malaria
SCD is most prevalent in areas in which malaria has historically been endemic. The sickle cell trait provides a carrier with a survival advantage against malaria fatality over people with normal haemoglobin in regions where malaria is endemic.[70][71]
Infection with the malaria parasite affects asymptomatic carriers of the abnormal haemoglobin gene differently from people with full sickle cell disease. Carriers (heterozygous for the gene) who catch malaria are less likely to suffer from severe symptoms than people with normal haemoglobin. People with sickle cell disease (homozygous for the gene) are similarly less likely to become infected with malaria; however, once infected, they are more likely to develop severe and life-threatening anaemia.[72][73]
The impact of sickle cell anaemia on malaria immunity illustrates some evolutionary trade-offs that have occurred because of endemic malaria. Although the shorter life expectancy for those with the homozygous condition would tend to disfavour the trait's survival, the trait is preserved in malaria-prone regions because of the benefits provided by the heterozygous form; an example of natural selection.[74]
Due to the adaptive advantage of the heterozygote, the disease is still prevalent, especially among people with recent ancestry in malaria-stricken areas, such as Africa, the Mediterranean, India, and the Middle East.[75] Malaria was historically endemic to southern Europe, but it was declared eradicated in the mid-20th century, except rare sporadic cases.[76]
The malaria parasite has a complex lifecycle and spends part of it in red blood cells. There are two mechanisms that protect sickle cell carriers from malaria. One is that the parasite is hindered from growing and reproducing in a carrier's red blood cells; another is that a carrier's red cells show signs of damage when infected, and are detected and destroyed as they pass through the spleen.[77][78]
Pathophysiology
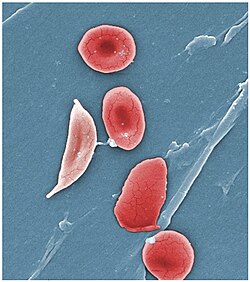
Scanning electron micrograph showing a mixture of red blood cells, some with round normal morphology, some with mild sickling showing elongation and bending
Under conditions of low oxygen concentration, haemoglobin S polymerises to form long strands within the red blood cell (RBC). These strands distort the shape of the cell and, after a few seconds, cause it to adopt an abnormal, inflexible, sickle-like shape. This process reverses when oxygen concentration is raised and the cells resume their normal biconcave disc shape. If sickling takes place in the venous system, after blood has passed through the capillaries, it does not affect the organs, and the RBCs can unsickle when they become oxygenated in the lungs. Repeated switching between sickle and normal shapes damages the membrane of the RBC so that it eventually becomes permanently sickled.[79][80][81]
Normal red blood cells are quite elastic and have a biconcave disc shape, which allows the cells to deform to pass through capillaries. In sickle cell disease, low oxygen tension promotes red blood cell sickling and repeated episodes of sickling damage the cell membrane and decrease the cell's elasticity. These cells fail to return to normal shape when normal oxygen tension is restored. As a consequence, these rigid blood cells are unable to deform as they pass through narrow capillaries, leading to vessel occlusion and ischaemia.[30]
Cells that have become sickled are detected as they pass through the spleen and are destroyed. In young children with sickle cell disease, the accumulation of sickled cells in the spleen can result in splenic sequestration crisis. In this, the spleen becomes engorged with blood, depriving the general circulation of blood cells and leading to severe anaemia. The spleen initially becomes noticeably swollen, but the lack of a healthy blood flow through the organ culminates in scarring of the spleen tissues and eventually death of the organ, generally before the age of 5 years.[80][31]
The actual anaemia of the illness is caused by haemolysis, the destruction of the red cells, because of their shape. Although the bone marrow attempts to compensate by creating new red cells, it does not match the rate of destruction. Healthy red blood cells typically function for 90–120 days, but sickled cells only last 10–20 days.[82][80]
The rapid breakdown of RBCs in sickle cell disease results in the release of free heme into the bloodstream, exceeding the capacity of the body's protective mechanisms. Although heme is an essential component of haemoglobin, it is also a potent oxidative molecule. Free heme is also an alarmin – a signal of tissue damage or infection, which triggers defensive responses in the body and increases the risk of inflammation and vaso-occlusive events.[83][84]
Diagnosis
Prenatal and newborn screening
Checking for sickle cell disease begins during pregnancy, with a prenatal screening questionnaire which includes, among other things, a consideration of health issues in the child's parents and close relatives. During pregnancy, genetic testing can be done on either a blood sample from the foetus or a sample of amniotic fluid. During the first trimester of pregnancy, chorionic villus sampling (CVS) is also a technique used for sickle cell disease prenatal diagnosis.[85] A routine heel prick test, in which a small sample of blood is collected a few days after birth, is used to check conclusively for sickle cell disease as well as other inherited conditions.[86]
Tests
A schematic of haemoglobin electrophoresis, showing the banding which is typical of various types of haemoglobin. Note that sickle cell disease (SCD) gives a single, bold band whereas sickle cell trait gives two slightly fainter bands.
Where sickle cell disease is suspected, a number of tests can be used. Often, a simpler, cheaper test is applied first, with a more complex test, such as DNA analysis, used to confirm a positive result.[87]
Two tests that are specific for sickle cell disease:
A blood smear is a thin layer of blood smeared on a glass microscope slide and then stained in such a way as to allow the various blood cells to be examined microscopically. This technique can be used to visually detect sickled cells; however, it does not detect sickle cell carriers.[87]
A solubility test relies on the fact that HbS is less soluble than normal haemoglobin (HbA); it is highly reliable but does not distinguish between full sickle cell disease and carrier status.[88]
Tests which can be used for sickle cell disease as well as for other hemoglobinopathies:
Haemoglobin electrophoresis is a test that can detect different types of haemoglobin. Haemoglobin is extracted from the red cells, then introduced into a porous gel and subjected to an electrical field. This separates the normal and abnormal types of haemoglobin, which can then be identified and quantified.[89]
Isoelectric focusing (IEF) is a technique that can be used to diagnose sickle cell disease and other hemoglobinopathies. The technique separates molecules based on their isoelectric point, or the pH at which they have no net electrical charge. IEF uses an electric charge to separate and identify different types of haemoglobin, which become focused into sharp, stationary bands. The technique can distinguish many types of abnormal haemoglobin.[90]
High-performance liquid chromatography (HPLC) is reliable, fully automated, and able to distinguish most types of sickle cell disease, including heterozygous. The method separates and quantifies haemoglobin fractions by measuring their rate of flow through a column of absorbent material.[87]
Genetic counselling is the process by which people with a hereditary disorder are advised of the probability of transmitting it and how this may be prevented or ameliorated.[91]
People who are known carriers of the disease or at risk of having a child with sickle cell anaemia may undergo genetic counselling. Genetic counsellors work with families to discuss the benefits, limitations, and logistics of genetic testing options as well as the potential impact of testing and test results on the individual.[92] Counselling is best given before a child is conceived, and several possible courses could be suggested. These include adoption, the use of eggs or sperm from a healthy donor, and in-vitro fertilisation (IVF) when combined with pre-implantation genetic diagnosis of the embryos.[93]
Several precautions can help reduce the risk of developing a sickling crisis. Lifestyle behaviours include maintaining good hydration and avoiding physical stress or exhaustion. Since sickling can be triggered by low oxygen levels, people with sickle cell disease should avoid high altitudes such as high mountains or flying in unpressurised aircraft.[94][95] People with sickle cell disease should avoid alcohol and smoking, as alcohol can cause dehydration and smoking can trigger acute chest syndrome. Stress can also trigger a sickle cell crisis, so relaxation techniques like breathing exercises can help.[95]
Pneumococcal infection is a leading cause of death among children with sickle cell disease; penicillin is recommended daily during the first 5 years of life to minimise the risk of infection.[96][97]
Dietary supplementation of folic acid is sometimes recommended, on the basis that it facilitates the creation of new red blood cells and may reduce anaemia. A Cochrane review of its use in 2016 found "the effect of supplementation on anaemia and any symptoms of anaemia remains unclear" due to a lack of medical evidence.[98][99]
People with sickle cell disease are recommended to receive all vaccinations which are recommended by health authorities to avoid serious infection, which might trigger a sickling crisis.[100][101]
Hydroxyurea was the first approved drug for the treatment of sickle cell disease, which has been shown to decrease the number and severity of attacks and possibly increase survival time.[102][103] This is achieved, in part, by reactivating foetal haemoglobin production in place of the haemoglobin S that causes sickling. Hydroxyurea lowers the expression of adhesion molecules on endothelial and red blood cells, which lowers the chance of small vessel blockages. Additionally, it encourages the release of nitric oxide, which enhances blood flow and inhibits the formation of clots.[104] Hydroxyurea had previously been used as a chemotherapy agent, and some concern exists that long-term use may be harmful.[25][105] A Cochrane review in 2022 found a weak evidence base for its use in sickle cell disease.[106]
Voxelotor was received accelerated approval as a treatment for sickle cell disease in the United States in 2019, and was approved by the European Medicines Agency (EMA) in 2021.[107] In trials, it had been shown to have disease-modifying potential by increasing haemoglobin levels and decreasing hemolysis indicators[108][109] However, following an increased risk of vaso-occlusive seizures and death observed in registries and clinical trials, the manufacturer, Pfizer, withdrew it from the market worldwide.[110][111]
The simple, or top-up transfusion, is a procedure in which healthy blood cells from a donor are infused into the patient's bloodstream. This benefits by alleviating anaemia and increasing oxygen levels in the tissues, reducing the risk of sickling and relieving sickling symptoms. A simple transfusion can be used to treat sickle cell disease when haemoglobin levels drop too low, or to prepare for an operation or pregnancy. It can also be used to protect against long-term complications or to reduce the risk of stroke.[112][113]
An exchange transfusion is a procedure in which blood is removed from the body, then processed to extract sickled cells, which are replaced by healthy red blood cells from a donor. The treated blood, including white cells and plasma, is then returned to the patient. Exchange transfusions are likely to be needed in an emergency, in severe cases of sickle cell disease, or to support a mother during pregnancy.[114]
Stroke prevention
Transcranial Doppler ultrasound (TCD) can detect children with sickle cell who have a high risk for stroke. The ultrasound test detects blood vessels partially obstructed by sickle cells by measuring the rate of blood into the brain, as blood flow velocity is inversely related to arterial diameter, and consequently, high blood flow velocity is correlated with narrowing of the arteries.[115]
In children, preventive RBC transfusion therapy has been shown to reduce the risk of first stroke or silent stroke when transcranial Doppler ultrasonography shows abnormal cerebral blood flow.[8] In those who have sustained a prior stroke event, it also reduces the risk of recurrent stroke and additional silent strokes.[116][117]
Vaso-occlusive crisis
Most people with sickle cell disease have intensely painful episodes called vaso-occlusive crises (VOC). However, the frequency, severity, and duration of these crises vary tremendously. In a VOC, the circulation of blood vessels is obstructed by sickled red blood cells, causing ischemic injuries to the tissues, inflammation, and pain. Recurrent episodes may cause irreversible organ damage.[118][119]
The most common and obvious symptom of a VOC is pain, which may be felt anywhere in the body but most commonly in the limbs and back. The degree of pain varies from mild to severe.[118] Home treatment options include bed rest and hydration, and pain control using over-the-counter medication such as paracetamol or ibuprofen.[120] More severe cases may require prescription opioids such as codeine or morphine for pain control.[121]
In 2019, crizanlizumab, a monoclonal antibody targeting P-selectin, was approved in the United States to reduce the frequency of vaso-occlusive crisis in those 16 years and older.[122] It had also been approved in the UK and the European Union, but in both cases authorisation was subsequently withdrawn because of poor evidence of its effectiveness.[123][124]
Acute chest syndrome
Acute chest syndrome is caused by vaso-occlusion occurring in the lungs. As with a VOC, treatment includes pain control and hydration. Antibiotics are required because there is a severe risk of pulmonary infection, and oxygen supplementation for hypoxia. Blood transfusion may also be required, or exchange transfusion in severe cases.[125]
Hematopoietic stem cells (HSC) are cells in the bone marrow that can develop into all types of blood cells, including red blood cells, white blood cells, and platelets.[127] There are two possible ways to treat sickle cell disease and some other hemoglobinopathies by targeting HSCs. Since 1991, a small number of patients have received bone marrow transplants from healthy matched donors, although this procedure has a high level of risk.[128] More recently, it has become possible to use CRISPR gene editing technology to modify the patient's own HSCs in a way that reduces or eliminates the production of sickle haemoglobin HbS and replaces it with a non-sickling form of haemoglobin.[129]
All stem cell treatments must involve myeloablation of the patients' bone marrow to remove HSCs containing the faulty gene. This requires high doses of chemotherapy agents with side effects such as sickness and tiredness. A long hospital stay is necessary after infusion of the replacement HSCs, while the cells take up residence in the bone marrow and start to make red blood cells with the stable form of haemoglobin.[130][131]
Gene therapy
Gene therapy was first trialled in 2014 on a single patient,[132] and followed by clinical trials in which several patients were successfully treated.[133] In 2023, both exagamglogene autotemcel (Casgevy) and lovotibeglogene autotemcel (Lyfgenia) were approved for the treatment of sickle cell disease.[15][131][17] Kendric Cromer in October 2024 became the first commercial case in the US to receive gene therapy and was discharged from Children's National Hospital.[134] The one-off gene-editing therapy, Casgevy, also known as Exa-cel, is to be offered to patients on the National Health Service (NHS) in England as from 2025.[135]
Both Casgevy and Lyfgenia work by first harvesting the patient's HSCs, then using CRISPR gene editing to modify their DNA in the laboratory. In parallel with this, the person with sickle cell disease's bone marrow is put through a myeloablation procedure to destroy the remaining HSCs. The treated cells are then infused back into the patient, where they colonise the bone marrow and eventually resume production of blood cells. Casgevy works by editing the BCL11A gene, which normally inhibits the production of haemoglobin F (foetal haemoglobin) in adults. The edit has the effect of increasing the production of HbF, which is not prone to sickling.[136] Lyfgenia introduces a new gene for T87Q-globin, which coexists with the sickling beta-globin but reduces the incidence of sickling.[137]
Hematopoietic stem cell transplantation
Hematopoietic stem cell transplantation (HSCT) involves replacing the dysfunctional stem cells from a person with sickle cell disease with healthy cells from a well-matched donor.[138][128] Finding a well matched donor is essential to the process' success. Different types of donors may be suitable and include umbilical cord blood, human leukocyte antigen (HLA) matched relatives, or HLA matched donors that are not related to the person being treated.[138] Risks associated with HSCT can include graft-versus-host disease, failure of the graft, and other toxicity related to the transplant.[138]
In contrast, life expectancy in the United States in 2010–2020 was 43 years[140] and in the UK 67 years.[12]
Epidemiology
The HbS gene can be found in every ethnic group.[141] The highest frequency of sickle cell disease is found in tropical regions, particularly sub-Saharan Africa, tribal regions of India, and the Middle East.[142] About 80% of sickle cell disease cases are believed to occur in Sub-Saharan Africa.[19] Migration of substantial populations from these high-prevalence areas to low-prevalence countries in Europe has dramatically increased in recent decades and in some European countries, sickle cell disease has now overtaken more familiar genetic conditions such as haemophilia and cystic fibrosis.[143] In 2015, it resulted in about 114,800 deaths.[144]
Sickle cell disease occurs more commonly among people whose ancestors lived in tropical and subtropical sub-Saharan regions where malaria is or was common. Where malaria is common, carrying a single sickle cell allele (trait) confers a heterozygote advantage; humans with one of the two alleles of sickle cell disease show less severe symptoms when infected with malaria.[145]
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.[24]
Africa
Three-quarters of sickle cell cases occur in Africa. A World Health Organization report dated 2006 estimated that around 2% of newborns in Nigeria are affected by sickle cell anaemia, giving a total of 150,000 affected children born every year in Nigeria alone. The carrier frequency ranges between 10 and 40% across equatorial Africa, decreasing to 1–2% on the North African coast and <1% in South Africa. In the West African countries of Ghana and Nigeria, the frequencies can vary from 15 to 30%. In Nigeria, 24% of the population carries the gene, and 20 per 1,000 newborns are born with the disease, or 150,000 annually.[146]
Uganda has the fifth-highest sickle cell disease burden in Africa.[147] One study indicates that 20,000 babies per year, or 0.7% of the total, are born with sickle cell disease, and 13.3% carry the trait. In Uganda, carrier frequency of the trait varies strongly across tribal lines: among the Baamba, it reaches 45%.[148]
United States
The number of people with the disease in the United States is about 100,000 (one in 3,300), mostly affecting Americans of sub-Saharan African descent.[149] In the United States, about one out of 365 African-American children and one in every 16,300 Hispanic-American children have sickle cell anaemia.[150] The life expectancy for men with sickle cell disease is approximately 42 years of age while women live approximately six years longer.[151] An additional 2million are carriers of the sickle cell trait.[152] Most infants with sickle cell disease born in the United States are identified by routine neonatal screening. As of 2016, all 50 states include screening for sickle cell disease as part of their newborn screening.[153] The newborn's blood is sampled through a heel-prick and is sent to a lab for testing. The baby must have been eating for a minimum of 24 hours before the heel-prick test can be done. Some states also require a second blood test to be done when the baby is two weeks old to ensure the results.[154]
Sickle cell disease is the most common genetic disorder among African Americans. Approximately 8% are carriers and 1 in 375 are born with the disease.[155] Patient advocates for sickle cell disease have complained that it gets less government and private research funding than similar rare diseases such as cystic fibrosis, with researcher Elliott Vichinsky saying this shows racial discrimination or the role of wealth in health care advocacy.[156] Overall, without considering race, approximately 1.5% of infants born in the United States are carriers of at least one copy of the mutant (disease-causing) gene.[157]
France
Percentage of newborns screened for sickle cell disease within mainland France from 2006 to 2018
As a result of population growth in African-Caribbean regions of overseas France and immigration from North and sub-Saharan Africa to mainland France, sickle cell disease has become a major health problem in France.[158] Sickle cell disease has become the most common genetic disease in the country, with an overall birth prevalence of one in 2,415 in mainland France, ahead of phenylketonuria (one in 10,862), congenital hypothyroidism (one in 3,132), congenital adrenal hyperplasia (one in 19,008) and cystic fibrosis (one in 5,014) for the same reference period.[159]
Percentage of newborns screened regionally and overall for sickle cell disease in mainland France and fraction of positive outcomes in all screened in 2018
Since 2000, neonatal screening of sickle cell disease has been performed at the national level for all newborns defined as being "at-risk" for sickle cell disease based on ethnic origin (defined as those born to parents originating from sub-Saharan Africa, North Africa, the Mediterranean area (South Italy, Greece, and Turkey), the Arabic peninsula, the French overseas islands, and the Indian subcontinent).[160] Since 3 August 2024, this screening is systematically applied to all newborns in France.[161]
United Kingdom
In the United Kingdom, between 12,000 and 15,000 people are thought to have sickle cell disease [162] with an estimated 250,000 carriers of the condition in England alone. As the number of carriers is only estimated, all newborn babies in the UK receive a routine blood test to screen for the condition.[163] Due to many adults in high-risk groups not knowing if they are carriers, pregnant women and both partners in a couple are offered screening so they can get counselling if they have the sickle cell trait.[164] In addition, blood donors from those in high-risk groups are also screened to confirm whether they are carriers and whether their blood filters properly.[165] Donors who are found to be carriers are informed and their blood, while often used for those of the same ethnic group, is not used for those with sickle cell disease who require a blood transfusion.[166]
West Asia
In Saudi Arabia, about 4.2% of the population carries the sickle cell trait, and 0.26% have sickle cell disease. The highest prevalence is in the Eastern province, where approximately 17% of the population carries the gene and 1.2% have sickle cell disease.[167] In 2005, Saudi Arabia introduced a mandatory premarital test including HB electrophoresis, which aimed to decrease the incidence of sickle cell disease and thalassemia.[168]
In Bahrain, a study published in 1998 that covered about 56,000 people in hospitals in Bahrain found that 2% of newborns have sickle cell disease, 18% of the surveyed people have the sickle cell trait, and 24% were carriers of the gene mutation causing the disease.[169] The country began screening of all pregnant women in 1992, and newborns started being tested if the mother was a carrier. In 2004, a law was passed requiring couples planning to marry to undergo free premarital counselling. These programmes were accompanied by public education campaigns.[170]
India and Nepal
Sickle cell disease is common in some ethnic groups of central India,[171] where the prevalence has ranged from 9.4 to 22.2% in endemic areas of Madhya Pradesh, Rajasthan, and Chhattisgarh.[172] It is also endemic among Tharu people of Nepal and India; however, they have a sevenfold lower rate of malaria despite living in a malaria infested zone.[173]
Caribbean Islands
In Jamaica, 10% of the population carries the sickle cell gene, making it the most prevalent genetic disorder in the country.[174]
History
The first modern report of sickle cell disease may have been in 1846, where the autopsy of an executed runaway slave was discussed; the key finding was the absence of the spleen.[175][176] Reportedly, African slaves in the United States exhibited resistance to malaria, but were prone to leg ulcers.[176] The abnormal characteristics of the red blood cells, which later lent their name to the condition, was first described by Ernest E. Irons (1877–1959), intern to Chicago cardiologist and professor of medicine James B. Herrick (1861–1954), in 1910. Irons saw "peculiar elongated and sickle-shaped" cells in the blood of a man named Walter Clement Noel, a 20-year-old first-year dental student from Grenada. Noel had been admitted to the Chicago Presbyterian Hospital in December 1904 with anaemia.[21][177] Noel was readmitted several times over the next three years for "muscular rheumatism" and "bilious attacks" but completed his studies and returned to the capital of Grenada (St. George's) to practice dentistry. He died of pneumonia in 1916 and is buried in the Catholic cemetery at Sauteurs in the north of Grenada.[21][22] Shortly after the report by Herrick, another case appeared in the Virginia Medical Semi-Monthly with the same title, "Peculiar Elongated and Sickle-Shaped Red Blood Corpuscles in a Case of Severe Anemia."[178] This article is based on a patient admitted to the University of Virginia Hospital on 15 November 1910.[179] In the later description by Verne Mason in 1922, the name "sickle cell anemia" is first used.[22][180] Childhood problems related to sickle cells disease were not reported until the 1930s, despite the fact that this cannot have been uncommon in African-American populations.[176]
Memphis physician Lemuel Diggs, a prolific researcher into sickle cell disease, first introduced the distinction between sickle cell disease and trait in 1933, although until 1949, the genetic characteristics had not been elucidated by James V. Neel and E.A. Beet.[22] 1949 was the year when Linus Pauling described the unusual chemical behaviour of haemoglobin S, and attributed this to an abnormality in the molecule itself.[22][181] The molecular change in HbS was described in 1956 by Vernon Ingram.[182] The late 1940s and early 1950s saw further understanding in the link between malaria and sickle cell disease. In 1954, the introduction of haemoglobin electrophoresis allowed the discovery of particular subtypes, such as HbSC disease.[22]
Large-scale natural history studies and further intervention studies were introduced in the 1970s and 1980s, leading to widespread use of prophylaxis against pneumococcal infections among other interventions. Bill Cosby's Emmy-winning 1972 TV movie, To All My Friends on Shore, depicted the story of the parents of a child with sickle cell disease.[183] The 1990s had the development of hydroxycarbamide, and reports of cure through bone marrow transplantation appeared in 2007.[22]
In the US, there can be stigma that hinders people with sickle cell disease from receiving necessary care;[185] one element of this is attributed to racism as the majority of people with sickle cell disease are black.[186] Due to this, in 1970, the Black Panther Party (also known as the Black Panther Party for Self Defense) opened dozens of free clinics across the U.S where free Sickle Cell Disease tests where offered. Over the course of the 1970s, thousands of people, largely African-Americans, received testing at one of these clinics.[187]
In September 2017, the US Social Security Administration issued a policy interpretation ruling providing background information on sickle cell disease and a description of how Social Security evaluates the disease during its adjudication process for disability claims.[188][189]
Uganda
Uganda has the fifth highest sickle cell disease (SCD) burden in the world.[190] In Uganda, social stigma exists for those with sickle cell disease because of the lack of general knowledge of the disease. The general gap in knowledge surrounding sickle cell disease is noted among adolescents and young adults due to the culturally sanctioned secrecy about the disease.[190] While most people have heard generally about the disease, a large portion of the population is relatively misinformed about how sickle cell disease is diagnosed or inherited. Those who are informed about the disease learned about it from family or friends and not from health professionals. Failure to provide the public with information about sickle cell disease results in a population with a poor understanding of the causes of the disease, symptoms, and prevention techniques.[147] The differences, physically and socially, that arise in those with sickle cell disease, such as jaundice, stunted physical growth, and delayed sexual maturity, can also lead them to become targets of bullying, rejection, and stigma.[190]
Rate of sickle cell disease in Uganda
The data compiled on sickle cell disease in Uganda has not been updated since the early 1970s. The deficiency of data is due to a lack of government research funds, even though Ugandans die daily from sickle cell disease.[191] Data shows that the trait frequency of sickle cell disease is 20% of the population in Uganda.[191] It is also estimated that about 25,000 Ugandans are born each year with sickle cell disease and 80% of those people do not live past five years old.[191] Sickle cell disease also contributes 25% to the child mortality rate in Uganda.[191] The Bamba people of Uganda, located in the southwest of the country, carry 45% of the gene which is the highest trait frequency recorded in the world.[191] The Sickle Cell Clinic in Mulago is only one sickle cell disease clinic in the country and, on average, sees 200 patients a day.[191]
Misconceptions about sickle cell disease
The stigma around the disease is particularly bad in regions of the country that are not as affected. For example, Eastern Ugandans tend to be more knowledgeable of the disease than Western Ugandans, who are more likely to believe that sickle cell disease resulted as a punishment from God or witchcraft.[192] Other misconceptions about sickle cell disease include the belief that it is caused by environmental factors but, in reality, sickle cell disease is a genetic disease.[193] There have been efforts throughout Uganda to address the social misconceptions about the disease. In 2013, the Uganda Sickle Cell Rescue Foundation was established to spread awareness of sickle cell disease and combat the social stigma attached to the disease.[194] In addition to this organisation's efforts, there is a need for the inclusion of sickle cell disease education in preexisting community health education programmes in order to reduce the stigmatisation of sickle cell disease in Uganda.[147]
Social isolation of people with sickle cell disease
The deeply rooted stigma of sickle cell disease in society causes families to often hide their family members' sick status for fear of being labeled, cursed, or left out of social events.[195] Sometimes in Uganda, when it is confirmed that a family member has sickle cell disease, intimate relationships with all members of the family are avoided.[195] The stigmatisation and social isolation that people with sickle cell disease tend to experience are often the consequence of popular misconceptions that people with sickle cell disease should not socialise with those free from the disease. This mentality robs people with sickle cell disease of the right to freely participate in community activities like everyone else.[190] SCD-related stigma and social isolation in schools, especially, can make life for young people living with sickle cell disease challenging.[190] For school-aged children living with sickle cell disease, the stigma they face can lead to peer rejection.[190] Peer rejection involves the exclusion from social groups or gatherings. It often leads the excluded individual to experience emotional distress and may result in their academic underperformance, avoidance of school, and occupational failure later in life.[190] This social isolation is also likely to negatively impact people with sickle cell disease's self-esteem and overall quality of life.[190]
Mothers of children with sickle cell disease tend to receive disproportionate amounts of stigma from their peers and family members. These women will often be blamed for their child's diagnosis of sickle cell disease, especially if sickle cell disease is not present in earlier generations, due to the suspicion that the child's poor health may have been caused by the mother's failure to implement preventative health measures or promote a healthy environment for her child to thrive.[193] The reliance on theories related to environmental factors to place blame on the mother reflects many Ugandans' poor knowledge of how the disease is acquired as it is determined by genetics, not environment.[193] Mothers of children with sickle cell disease are also often left with very limited resources to safeguard their futures against the stigma of having sickle cell disease.[193] This lack of access to resources results from their subordinating roles within familial structures as well as the class disparities that hinder many mothers' ability to satisfy additional childcare costs and responsibilities.[193]
Women living with sickle cell disease who become pregnant often face extreme discrimination and discouragement in Uganda. These women are frequently branded by their peers as irresponsible for having a baby while living with sickle cell disease or even engaging in sex while living with sickle cell disease.[196] The criticism and judgement these women receive, not only from healthcare professionals but also from their families, often leaves them feeling alone, depressed, anxious, ashamed, and with very little social support.[197] Most pregnant women with sickle cell disease also go on to be single mothers as it is common for them to be left by their male partners who claim they were unaware of their partner's sickle cell disease status.[198] Not only does the abandonment experienced by these women cause emotional distress for them, but this low level of parental support can be linked to depressive symptoms and overall lower quality of life for the child once they are born.[199]
United Kingdom
The National Health Service (NHS) makes a number of treatments available for people with sickle cell disease. These include pain management, antibiotics and vaccinations to reduce the risk of infection, and blood transfusions where appropriate.[200] Other treatments include Voxelotor which reduces the need for blood transfusions,[201] and the gene therapy Casgevy.[202] In England, a number of regional centres coordinate treatment for sickle cell disease and other haemoglobinopathies.[203]
Media and arts representation of sickle cell disease
Popular media and art have been important educational tools about sickle cell disease.[204]Hertz Nazaire was a Haitian-American visual artist, writer, and advocate who lived with sickle cell disease and used his art to raise awareness, combat stigma, and champion better access to care.[205]
Representations of sickle cell disease in television shows include in the longrunning American medical drama ER (season 4 "Obstruction of Justice" and season 15 "Separation Anxiety" and "Dream Runner"), the 2024 British superhero series Supacell, in the 2024 period drama Lady in the Lake, and in the 2025 medicalprocedural dramaThe Pitt (episode "8:00 am"). Noah Wyle, who plays Dr. Michael "Robby" Robinavitch on The Pitt, has spoken about the importance of popular media representation of sickle cell and the impacts of racism on the quality of care patients receive.[206]
12Nelson MD, Bennett DM, Lehman ME, Okonji AI (December 2022). "Dizziness, Falls, and Hearing Loss in Adults Living With Sickle Cell Disease". American Journal of Audiology. 31 (4): 1178–1190. doi:10.1044/2022_AJA-22-00059. ISSN1558-9137. PMID36251873.
123Savitt TL, Goldberg MF (January 1989). "Herrick's 1910 case report of sickle cell anemia. The rest of the story". The Journal of the American Medical Association. 261 (2): 266–271. doi:10.1001/jama.261.2.266. PMID2642320.
123"Sickle cell disease". MedlinePlus Genetics. United States: National Library of Medicine. Retrieved 13 November 2024.
12Yawn BP, Buchanan GR, Afenyi-Annan AN, Ballas SK, Hassell KL, James AH, etal. (September 2014). "Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members". The Journal of the American Medical Association. 312 (10): 1033–1048. doi:10.1001/jama.2014.10517. PMID25203083. S2CID37681044.
12Gill FM, Sleeper LA, Weiner SJ, Brown AK, Bellevue R, Grover R, etal. (July 1995). "Clinical events in the first decade in a cohort of infants with sickle cell disease. Cooperative Study of Sickle Cell Disease [see comments]". Blood. 86 (2): 776–783. doi:10.1182/blood.v86.2.776.bloodjournal862776. ISSN0006-4971.
↑Friend A, Settelmeyer TP, Girzadas D (25 November 2023). "Acute Chest Syndrome". StatPearls. Treasure Island, Florida. PMID28722902. Retrieved 27 October 2024.
↑Kumar V, Abbas AK, Fausto N, Aster J (28 May 2009). Robbins and Cotran Pathologic Basis of Disease (Professionaled.). Elsevier Health. pp.Kindle Location 33329.
↑Kavanagh PL, Sprinz PG, Vinci SR, Bauchner H, Wang CJ (December 2011). "Management of children with sickle cell disease: a comprehensive review of the literature". Pediatrics. 128 (6): e1552–e1574. doi:10.1542/peds.2010-3686. PMID22123880. S2CID14524078.
↑Chrouser KL, Ajiboye OB, Oyetunji TA, Chang DC (April 2011). "Priapism in the United States: the changing role of sickle cell disease". American Journal of Surgery. 201 (4): 468–474. doi:10.1016/j.amjsurg.2010.03.017. PMID21421100.
↑Elagouz M, Jyothi S, Gupta B, Sivaprasad S (July 2010). "Sickle cell disease and the eye: old and new concepts". Survey of Ophthalmology. 55 (4): 359–377. doi:10.1016/j.survophthal.2009.11.004. PMID20452638.
↑Rai P, Niss O, Malik P (November 2017). "A reappraisal of the mechanisms underlying the cardiac complications of sickle cell anemia". Pediatric Blood & Cancer. 64 (11) e26607. doi:10.1002/pbc.26607. PMID28453224. S2CID24444332.
↑Green NS, Fabry ME, Kaptue-Noche L, Nagel RL (October 1993). "Senegal haplotype is associated with higher HbF than Benin and Cameroon haplotypes in African children with sickle cell anemia". American Journal of Hematology. 44 (2): 145–146. doi:10.1002/ajh.2830440214. PMID7505527. S2CID27341091.
↑Padilla F, Bromberg PA, Jensen WN (May 1973). "The Sickle-Unsickle Cycle: A Cause of Cell Fragmentation Leading to Permanently Deformed Cells". Blood. 41 (5): 653–660. doi:10.1182/blood.V41.5.653.653. ISSN0006-4971. PMID4694082.
↑Colah, R. B., Gorakshakar, A. C., & Nadkarni, A. H. (2011). Invasive & non-invasive approaches for prenatal diagnosis of haemoglobinopathies: experiences from India. The Indian Journal of Medical Research, 134(4), 552–560.
↑Mirre E, Brousse V, Berteloot L, Lambot-Juhan K, Verlhac S, Boulat C, etal. (March 2010). "Feasibility and efficacy of chronic transfusion for stroke prevention in children with sickle cell disease". European Journal of Haematology. 84 (3): 259–265. doi:10.1111/j.1600-0609.2009.01379.x. PMID19912310. S2CID24316310.
↑Ribeil JA, Hacein-Bey-Abina S, Payen E, Magnani A, Semeraro M, Magrin E, etal. (March 2017). "Gene Therapy in a Patient with Sickle Cell Disease". New England Journal of Medicine. 376 (9): 848–855. doi:10.1056/NEJMoa1609677. ISSN0028-4793. PMID28249145.
↑Karkoska K, McGann PT (November 2023). "Changing Trends in Sickle Cell Disease-Related Mortality in the United States over Four Decades". Blood. 142 (Supplement 1): 925. doi:10.1182/blood-2023-177988. ISSN0006-4971.
↑Edwards QT, Seibert D, Macri C, Covington C, Tilghman J (November 2004). "Assessing ethnicity in preconception counseling: genetics—what nurse practitioners need to know". Journal of the American Academy of Nurse Practitioners. 16 (11): 472–480. doi:10.1111/j.1745-7599.2004.tb00426.x. PMID15617360. S2CID7644129.
↑Bardakdjian J, Wajcman H (September 2004). "[Epidemiology of sickle cell anemia]". La Revue du Praticien (in French). 54 (14): 1531–1533. PMID15558961.
↑Washburn RE (1911). "Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia". The Virginia Medical Semi-Monthly. 15 (21): 490–493.
↑Mason VR (1922). "Sickle cell anemia". The Journal of the American Medical Association. 79 (16): 1318–1320. doi:10.1001/jama.1922.02640160038012. Reprinted in Mason VR (October 1985). "Landmark article Oct. 14, 1922: Sickle cell anemia. By V.R. Mason". The Journal of the American Medical Association. 254 (14): 1955–1957. doi:10.1001/jama.254.14.1955. PMID3900438.
↑"Foster, Gloria". Facts On File History Database. Archived from the original on 5 March 2016. Retrieved 25 February 2015.
↑Richard-Lenoble D, Toublanc JE, Zinsou RD, Kombila M, Carme B (1980). "[Results of a systematic study of drepanocytosis in 1,500 Gabonese using haemoglobin electrophoresis]" [Results of a systematic study of drepanocytosis in 1,500 Gabonese using haemoglobin electrophoresis]. Bulletin de la Société de Pathologie Exotique et de ses Filiales (in French). 73 (2): 200–206. PMID7460122.
↑Edwards R (20 January 2022), The Park Bench (Animation, Short, Family), Zoie Watkins, Aflac Cancer and Blood Disorders Center, Lion Forge Animation, Magnetic Dreams, retrieved 28 June 2025
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.