Related Research Articles
An allele, or allelomorph, is a variant of the sequence of nucleotides at a particular location, or locus, on a DNA molecule.

Tay–Sachs disease is a genetic disorder that results in the destruction of nerve cells in the brain and spinal cord. The most common form is infantile Tay–Sachs disease, which becomes apparent around the age of three to six months of age, with the baby losing the ability to turn over, sit, or crawl. This is then followed by seizures, hearing loss, and inability to move, with death usually occurring by the age of three to five. Less commonly, the disease may occur later in childhood, adolescence, or adulthood. These forms tend to be less severe, but the juvenile form typically results in death by age 15.

In genetics, dominance is the phenomenon of one variant (allele) of a gene on a chromosome masking or overriding the effect of a different variant of the same gene on the other copy of the chromosome. The first variant is termed dominant and the second is called recessive. This state of having two different variants of the same gene on each chromosome is originally caused by a mutation in one of the genes, either new or inherited. The terms autosomal dominant or autosomal recessive are used to describe gene variants on non-sex chromosomes (autosomes) and their associated traits, while those on sex chromosomes (allosomes) are termed X-linked dominant, X-linked recessive or Y-linked; these have an inheritance and presentation pattern that depends on the sex of both the parent and the child. Since there is only one copy of the Y chromosome, Y-linked traits cannot be dominant or recessive. Additionally, there are other forms of dominance, such as incomplete dominance, in which a gene variant has a partial effect compared to when it is present on both chromosomes, and co-dominance, in which different variants on each chromosome both show their associated traits.

Hereditary haemochromatosis type 1 is a genetic disorder characterized by excessive intestinal absorption of dietary iron, resulting in a pathological increase in total body iron stores. Humans, like most animals, have no mechanism to regulate excess iron, simply losing a limited amount through various means like sweating or menstruating.
A heterozygote advantage describes the case in which the heterozygous genotype has a higher relative fitness than either the homozygous dominant or homozygous recessive genotype. Loci exhibiting heterozygote advantage are a small minority of loci. The specific case of heterozygote advantage due to a single locus is known as overdominance. Overdominance is a rare condition in genetics where the phenotype of the heterozygote lies outside of the phenotypical range of both homozygote parents, and heterozygous individuals have a higher fitness than homozygous individuals.

Pleiotropy occurs when one gene influences two or more seemingly unrelated phenotypic traits. Such a gene that exhibits multiple phenotypic expression is called a pleiotropic gene. Mutation in a pleiotropic gene may have an effect on several traits simultaneously, due to the gene coding for a product used by a myriad of cells or different targets that have the same signaling function.

Sandhoff disease is a lysosomal genetic, lipid storage disorder caused by the inherited deficiency to create functional beta-hexosaminidases A and B. These catabolic enzymes are needed to degrade the neuronal membrane components, ganglioside GM2, its derivative GA2, the glycolipid globoside in visceral tissues, and some oligosaccharides. Accumulation of these metabolites leads to a progressive destruction of the central nervous system and eventually to death. The rare autosomal recessive neurodegenerative disorder is clinically almost indistinguishable from Tay–Sachs disease, another genetic disorder that disrupts beta-hexosaminidases A and S. There are three subsets of Sandhoff disease based on when first symptoms appear: classic infantile, juvenile and adult late onset.

GM2-gangliosidosis, AB variant is a rare, autosomal recessive metabolic disorder that causes progressive destruction of nerve cells in the brain and spinal cord. It has a similar pathology to Sandhoff disease and Tay–Sachs disease. The three diseases are classified together as the GM2 gangliosidoses, because each disease represents a distinct molecular point of failure in the activation of the same enzyme, beta-hexosaminidase. AB variant is caused by a failure in the gene that makes an enzyme cofactor for beta-hexosaminidase, called the GM2 activator.
Hemoglobin A2 (HbA2) is a normal variant of hemoglobin A that consists of two alpha and two delta chains (α2δ2) and is found at low levels in normal human blood. Hemoglobin A2 may be increased in beta thalassemia or in people who are heterozygous for the beta thalassemia gene.
Hemoglobin C is an abnormal hemoglobin in which glutamic acid residue at the 6th position of the β-globin chain is replaced with a lysine residue due to a point mutation in the HBB gene. People with one copy of the gene for hemoglobin C do not experience symptoms, but can pass the abnormal gene on to their children. Those with two copies of the gene are said to have hemoglobin C disease and can experience mild anemia. It is possible for a person to have both the gene for hemoglobin S and the gene for hemoglobin C; this state is called hemoglobin SC disease, and is generally more severe than hemoglobin C disease, but milder than sickle cell anemia.

Human homeostatic iron regulator protein, also known as the HFE protein, is a transmembrane protein that in humans is encoded by the HFE gene. The HFE gene is located on short arm of chromosome 6 at location 6p22.2
The GM2 gangliosidoses are a group of three related genetic disorders that result from a deficiency of the enzyme beta-hexosaminidase. This enzyme catalyzes the biodegradation of fatty acid derivatives known as gangliosides. The diseases are better known by their individual names: Tay–Sachs disease, AB variant, and Sandhoff disease.
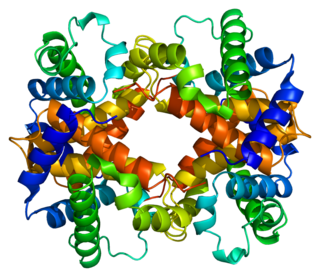
Hemoglobin subunit beta is a globin protein, coded for by the HBB gene, which along with alpha globin (HBA), makes up the most common form of haemoglobin in adult humans, hemoglobin A (HbA). It is 147 amino acids long and has a molecular weight of 15,867 Da. Normal adult human HbA is a heterotetramer consisting of two alpha chains and two beta chains.
Lethal alleles are alleles that cause the death of the organism that carries them. They are usually a result of mutations in genes that are essential for growth or development. Lethal alleles may be recessive, dominant, or conditional depending on the gene or genes involved.

Hexosaminidase A (alpha polypeptide), also known as HEXA, is an enzyme that in humans is encoded by the HEXA gene, located on the 15th chromosome.

Hemoglobin E (HbE) is an abnormal hemoglobin with a single point mutation in the β chain. At position 26 there is a change in the amino acid, from glutamic acid to lysine (E26K). Hemoglobin E is very common among people of Southeast Asian, Northeast Indian, Sri Lankan and Bangladeshi descent.
Haemochromatosis type 3 is a type of iron overload disorder associated with deficiencies in transferrin receptor 2. It exhibits an autosomal recessive inheritance pattern. The first confirmed case was diagnosed in 1865 by French doctor Trousseau. Later in 1889, the German doctor von Recklinghausen indicated that the liver contains iron, and due to bleeding being considered to be the cause, he called the pigment "Haemochromatosis." In 1935, English doctor Sheldon's groundbreaking book titled, Haemochromatosis, reviewed 311 patient case reports and presented the idea that haemochromatosis was a congenital metabolic disorder. Hereditary haemochromatosis is a congenital disorder which affects the regulation of iron metabolism thus causing increased gut absorption of iron and a gradual build-up of pathologic iron deposits in the liver and other internal organs, joint capsules and the skin. The iron overload could potentially cause serious disease from the age of 40–50 years. In the final stages of the disease, the major symptoms include liver cirrhosis, diabetes and bronze-colored skin. There are four types of hereditary hemochromatosis which are classified depending on the age of onset and other factors such as genetic cause and mode of inheritance.

Zygosity is the degree to which both copies of a chromosome or gene have the same genetic sequence. In other words, it is the degree of similarity of the alleles in an organism.
The medical genetics of Jews have been studied to identify and prevent some rare genetic diseases that, while still rare, are more common than average among people of Jewish descent. There are several autosomal recessive genetic disorders that are more common than average in ethnically Jewish populations, particularly Ashkenazi Jews, because of relatively recent population bottlenecks and because of consanguineous marriage. These two phenomena reduce genetic diversity and raise the chance that two parents will carry a mutation in the same gene and pass on both mutations to a child.

Hemoglobin Lepore syndrome is typically an asymptomatic hemoglobinopathy, which is caused by an autosomal recessive genetic mutation. The Hb Lepore variant, consisting of two normal alpha globin chains (HBA) and two delta-beta globin fusion chains which occurs due to a "crossover" between the delta (HBD) and beta globin (HBB) gene loci during meiosis and was first identified in the Lepore family, an Italian-American family, in 1958. There are three varieties of Hb Lepore, Washington, Baltimore and Hollandia. All three varieties show similar electrophoretic and chromatographic properties and hematological findings bear close resemblance to those of the beta-thalassemia trait; a blood disorder that reduces the production of the iron-containing protein hemoglobin which carries oxygen to cells and which may cause anemia.
References
- ↑ KJ Allen; LC Gurrin; CC Constantine; et al. (2008-01-17). "Iron-Overload–Related Disease in HFE Hereditary Hemochromatosis" (PDF). New England Journal of Medicine. 358 (3): 221–230. doi:10.1056/NEJMoa073286. PMID 18199861.
- ↑ Rossi E, Olynyk JK, Cullen DJ, Papadopoulos G, Bulsara M, Summerville L, Powell LW (Feb 2000). "Compound heterozygous hemochromatosis genotype predicts increased iron and erythrocyte indices in women". Clinical Chemistry. 46 (2): 162–166. doi:10.1093/clinchem/46.2.162. PMID 10657371.
- ↑ Deugnier Y, Mosser J (Aug 2008). "Modifying factors of the HFE hemochromatosis phenotype". Expert Review of Gastroenterology & Hepatology. 2 (4): 531–540. doi:10.1586/17474124.2.4.531. PMID 19072401.
- ↑ Anderson JA, Fisch R, Miller E, Doeden D (Mar 1966). "Atypical phenylketonuric heterozygote. Deficiency in phenylalanine hydroxylase and transaminase activity". Journal of Pediatrics. 68 (3): 351–360. doi:10.1016/s0022-3476(66)80237-8. PMID 4379218.
- ↑ Avigad S, Kleiman S, Weinstein M, Cohen BE, Schwartz G, Woo SL, Shiloh Y (Aug 1991). "Compound heterozygosity in nonphenylketonuria hyperphenylalanemia: The contribution of mutations for classical phenylketonuria". American Journal of Human Genetics. 49 (2): 393–399. PMC 1683284 . PMID 1867197.
- ↑ Ohno, Kousaku & Suzuki, Kunihiko (1988-12-05). "Multiple Abnormal beta-Hexosaminidase alpha-Chain mRNAs in a Compound-Heterozygous Ashkenazi Jewish Patient with Tay–Sachs Disease". Journal of Biological Chemistry. 263 (34): 18563–7. doi: 10.1016/S0021-9258(19)81396-0 . PMID 2973464.
- ↑ Gonzalez-Redondo JM, Stoming TA, Lanclos KD, et al. (1988). "Clinical and genetic heterogeneity in black patients with homozygous beta-thalassemia from the southeastern United States". Blood. 72 (3): 1007–1014. doi: 10.1182/blood.V72.3.1007.bloodjournal7231007 . PMID 2458145.
- ↑ Witkowska HE; Lubin BH; Beuzard Y; Baruchel S; Esseltine DW; Vichinsky EP; Kleman KM; Bardakdjian-Michau J; Pinkoski L; Cahn S; et al. (1991-10-17). "Sickle cell disease in a patient with sickle cell trait and compound heterozygosity for hemoglobin S and hemoglobin Quebec-Chori". New England Journal of Medicine. 325 (16). Massachusetts Medical Society: 1150–1154. doi: 10.1056/NEJM199110173251607 . PMID 1891024.