Related Research Articles

Hemoglobinopathy is the medical term for a group of inherited blood disorders and diseases that primarily affect red blood cells. They are single-gene disorders and, in most cases, they are inherited as autosomal co-dominant traits.

Thalassemias are inherited blood disorders that result in abnormal hemoglobin. Symptoms depend on the type of thalassemia and can vary from none to severe. Often there is mild to severe anemia as thalassemia can affect the production of red blood cells and also affect how long the red blood cells live. Symptoms of anemia include feeling tired and having pale skin. Other symptoms of thalassemia include bone problems, an enlarged spleen, yellowish skin, pulmonary hypertension, and dark urine. Slow growth may occur in children. Symptoms and presentations of thalassemia can change over time.
Fetal hemoglobin, or foetal haemoglobin is the main oxygen carrier protein in the human fetus. Hemoglobin F is found in fetal red blood cells, and is involved in transporting oxygen from the mother's bloodstream to organs and tissues in the fetus. It is produced at around 6 weeks of pregnancy and the levels remain high after birth until the baby is roughly 2–4 months old. Hemoglobin F has a different composition than adult forms of hemoglobin, allowing it to bind oxygen more strongly; this in turn enables the developing fetus to retrieve oxygen from the mother's bloodstream, which occurs through the placenta found in the mother's uterus.
An autosplenectomy is a negative outcome of disease and occurs when a disease damages the spleen to such an extent that it becomes shrunken and non-functional. The spleen is an important immunological organ that acts as a filter for red blood cells, triggers phagocytosis of invaders, and mounts an immunological response when necessary. Lack of a spleen, called asplenia, can occur by autosplenectomy or the surgical counterpart, splenectomy. Asplenia can increase susceptibility to infection. Autosplenectomy can occur in cases of sickle-cell disease where the misshapen cells block blood flow to the spleen, causing scarring and eventual atrophy of the organ. Autosplenectomy is a rare condition that is linked to certain diseases but is not a common occurrence. It is also seen in systemic lupus erythematosus (SLE).
Hemoglobin A (HbA), also known as adult hemoglobin, hemoglobin A1 or α2β2, is the most common human hemoglobin tetramer, accounting for over 97% of the total red blood cell hemoglobin. Hemoglobin is an oxygen-binding protein, found in erythrocytes, which transports oxygen from the lungs to the tissues. Hemoglobin A is the most common adult form of hemoglobin and exists as a tetramer containing two alpha subunits and two beta subunits (α2β2). Hemoglobin A2 (HbA2) is a less common adult form of hemoglobin and is composed of two alpha and two delta-globin subunits. This hemoglobin makes up 1-3% of hemoglobin in adults.
Hemoglobin A2 (HbA2) is a normal variant of hemoglobin A that consists of two alpha and two delta chains (α2δ2) and is found at low levels in normal human blood. Hemoglobin A2 may be increased in beta thalassemia or in people who are heterozygous for the beta thalassemia gene.
Hemoglobin C is an abnormal hemoglobin in which glutamic acid residue at the 6th position of the β-globin chain is replaced with a lysine residue due to a point mutation in the HBB gene. People with one copy of the gene for hemoglobin C do not experience symptoms, but can pass the abnormal gene on to their children. Those with two copies of the gene are said to have hemoglobin C disease and can experience mild anemia. It is possible for a person to have both the gene for hemoglobin S and the gene for hemoglobin C; this state is called hemoglobin SC disease, and is generally more severe than hemoglobin C disease, but milder than sickle cell anemia.

Alpha-thalassemia is a form of thalassemia involving the genes HBA1 and HBA2. Thalassemias are a group of inherited blood conditions which result in the impaired production of hemoglobin, the molecule that carries oxygen in the blood. Normal hemoglobin consists of two alpha chains and two beta chains; in alpha-thalassemia, there is a quantitative decrease in the amount of alpha chains, resulting in fewer normal hemoglobin molecules. Furthermore, alpha-thalassemia leads to the production of unstable beta globin molecules which cause increased red blood cell destruction. The degree of impairment is based on which clinical phenotype is present.
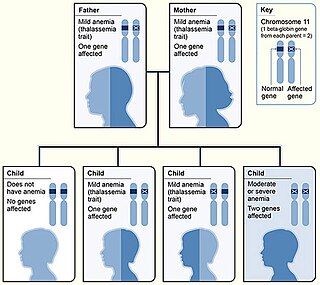
Beta thalassemias are a group of inherited blood disorders. They are forms of thalassemia caused by reduced or absent synthesis of the beta chains of hemoglobin that result in variable outcomes ranging from severe anemia to clinically asymptomatic individuals. Global annual incidence is estimated at one in 100,000. Beta thalassemias occur due to malfunctions in the hemoglobin subunit beta or HBB. The severity of the disease depends on the nature of the mutation.
Hemoglobin subunit beta is a globin protein, coded for by the HBB gene, which along with alpha globin (HBA), makes up the most common form of haemoglobin in adult humans, hemoglobin A (HbA). It is 147 amino acids long and has a molecular weight of 15,867 Da. Normal adult human HbA is a heterotetramer consisting of two alpha chains and two beta chains.

Hemoglobin variants are different types of hemoglobin molecules, by different combinations of its subunits and/or mutations thereof. Hemoglobin variants are a part of the normal embryonic and fetal development. They may also be pathologic mutant forms of hemoglobin in a population, caused by variations in genetics. Some well-known hemoglobin variants, such as sickle-cell anemia, are responsible for diseases and are considered hemoglobinopathies. Other variants cause no detectable pathology, and are thus considered non-pathological variants.
In medical genetics, compound heterozygosity is the condition of having two or more heterogeneous recessive alleles at a particular locus that can cause genetic disease in a heterozygous state; that is, an organism is a compound heterozygote when it has two recessive alleles for the same gene, but with those two alleles being different from each other. Compound heterozygosity reflects the diversity of the mutation base for many autosomal recessive genetic disorders; mutations in most disease-causing genes have arisen many times. This means that many cases of disease arise in individuals who have two unrelated alleles, who technically are heterozygotes, but both the alleles are defective.
Hemoglobin Barts, abbreviated Hb Barts, is an abnormal type of hemoglobin that consists of four gamma globins. It is moderately insoluble, and therefore accumulates in the red blood cells. Hb Barts has an extremely high affinity for oxygen, so it cannot release oxygen to the tissue. Therefore, this makes it an inefficient oxygen carrier. As an embryo develops, it begins to produce alpha-globins at weeks 5–6 of development. When both of the HBA1 and HBA2 genes which code for alpha globins becomes dysfunctional, the affected fetuses will have difficulty in synthesizing a functional hemoglobin. As a result, gamma chains will accumulate and form four gamma globins. These gamma globins bind to form hemoglobin Barts. It is produced in the disease alpha-thalassemia and in the most severe of cases, it is the only form of hemoglobin in circulation. In this situation, a fetus will develop hydrops fetalis and normally die before or shortly after birth, unless intrauterine blood transfusion is performed.

Hemoglobin subunit alpha, Hemoglobin, alpha 1, is a hemoglobin protein that in humans is encoded by the HBA1 gene.
Hereditary persistence of fetal hemoglobin (HPFH) is a benign condition in which increased fetal hemoglobin production continues well into adulthood, disregarding the normal shutoff point after which only adult-type hemoglobin should be produced.

Hemoglobin E (HbE) is an abnormal hemoglobin with a single point mutation in the β chain. At position 26 there is a change in the amino acid, from glutamic acid to lysine (E26K). Hemoglobin E is very common among people of Southeast Asian, Northeast Indian, Sri Lankan and Bangladeshi descent.

Hemoglobin, alpha 2 also known as HBA2 is a gene that in humans codes for the alpha globin chain of hemoglobin.
Human genetic resistance to malaria refers to inherited changes in the DNA of humans which increase resistance to malaria and result in increased survival of individuals with those genetic changes. The existence of these genotypes is likely due to evolutionary pressure exerted by parasites of the genus Plasmodium which cause malaria. Since malaria infects red blood cells, these genetic changes are most common alterations to molecules essential for red blood cell function, such as hemoglobin or other cellular proteins or enzymes of red blood cells. These alterations generally protect red blood cells from invasion by Plasmodium parasites or replication of parasites within the red blood cell.
Within the medical specialty of hematology, Hemoglobin D-Punjab, also known as hemoglobin D-Los Angeles, D-North Carolina, D-Portugal, D-Oak Ridge, and D-Chicago, is a hemoglobin variant. It originates from a point mutation in the human β-globin locus and is one of the most common hemoglobin variants worldwide. It is so named because of its higher prevalence in the Punjab region of India and Pakistan, along with northern China, and North America. It is also the most frequent hemoglobin variant in Xinjiang Uyghur Autonomous Region of China, with a 1997 study indicating that Hemoglobin D-Punjab accounts for 55.6% of the total hemoglobin variants.
Hemoglobin H disease, also called alpha-thalassemia intermedia, is a disease affecting hemoglobin, the oxygen carrying molecule within red blood cells. It is a form of Alpha-thalassemia which most commonly occurs due to deletion of 3 out of 4 of the α-globin genes.
References
- ↑ "Newborn Screening Program - Sickle Cell Beta Thalassemia Disease". www.idph.state.il.us. Retrieved 2015-06-18.
- ↑ Ashley-Koch, A; Yang, Q; Olney, R. S. (2000). "Sickle hemoglobin (HbS) allele and sickle cell disease: A HuGE review". American Journal of Epidemiology. 151 (9): 839–45. doi: 10.1093/oxfordjournals.aje.a010288 . PMID 10791557.
- ↑ "Hemoglobin S– β -Thalassemia Disease - Hematology and Oncology" . Retrieved 2015-06-18.
- ↑ "Newborn Screening Program - Sickle Cell Beta Thalassemia Disease". www.idph.state.il.us. Retrieved 2015-06-18.
- ↑ Ashley-Koch, A; Yang, Q; Olney, R. S. (2000). "Sickle hemoglobin (HbS) allele and sickle cell disease: A HuGE review". American Journal of Epidemiology. 151 (9): 839–45. doi: 10.1093/oxfordjournals.aje.a010288 . PMID 10791557.