There are two main groups: abnormal structural hemoglobin variants caused by mutations in the hemoglobin genes, and the thalassemias, which are caused by an underproduction of otherwise normal hemoglobin molecules. The main structural hemoglobin variants are HbS, HbE and HbC. The main types of thalassemia are alpha-thalassemia and beta thalassemia.[4][2]
Hemoglobin functions
Hemoglobin is a protein containing iron that facilitates the transportation of oxygen in red blood cells.[5] Hemoglobin in the blood carries oxygen from the lungs to the other tissues of the body, where it releases the oxygen to enable aerobic respiration which powers the metabolism. Normal levels of hemoglobin vary according to sex and age in the range 9.5 to 17.2 grams of hemoglobin in every deciliter of blood.[6]
Hemoglobin also transports other gases. It carries off some of the body's respiratory carbon dioxide (about 20–25% of the total)[7] as carbaminohemoglobin, in which CO2 binds to the heme protein. The molecule also carries the important regulatory molecule nitric oxide bound to a thiol group in the globin protein, releasing it at the same time as oxygen.[8]
Hemoglobin structural biology
(a) schematic representation of a hemoglobin molecule, showing alpha and beta globins. (b) structure of the heme molecular component of hemoglobin
Normal human hemoglobins are tetrameric proteins composed of two pairs of globin chains, each of which contains one alpha-like (α) globin and one beta-like (β) globin. Each globin chain is associated with an iron-containing hememoiety. Throughout life, the synthesis of the α and the β chains is balanced so that their ratio is relatively constant and there is no excess of either type.[9]
The specific α and β chains that are incorporated into Hb are highly regulated during development:[10]
Embryonic Hb are expressed as early as four to six weeks of embryogenesis and disappear around the eighth week of gestation as they are replaced by fetal Hb.[11][12] Embryonic Hbs include:
Hb Gower-1, composed of two ζ (zeta) globins and two ε (epsilon) globins, i.e., ζ2ε2
Hb Gower-2, composed of two α globins and two ε globins (α2ε2)
Hb Portland, composed of two ζ globins and two γ (gamma) globins (ζ2γ2)
Fetal Hb (HbF) is produced from approximately eight weeks of gestation through birth and constitutes approximately 80 percent of Hb in the full-term neonate. It declines during the first few months of life and, in the normal state, constitutes <1 percent of total Hb by early childhood. HbF is composed of two α globins and two γ globins (α2γ2).[13]
Adult Hb (HbA) is the predominant Hb in children by six months of age and onward; it constitutes 96-97% of total Hb in individuals without a hemoglobinopathy. It is composed of two α globins and two β globins (α2β2).[14]
HbA2 is a minor adult Hb that normally accounts for approximately 2.5–3.5% of total Hb from six months of age onward. It is composed of two α globins and two δ (delta) globins (α2δ2).[14]
Classification of hemoglobinopathies
A) Qualitative
Structural abnormalities
Hemoglobin structural variants manifest a change in the structure of the Hb molecule. The majority of hemoglobin variants do not cause disease and are most commonly discovered either incidentally or through newborn screening. Hb variants can usually be detected by protein-based assay methods such as electrophoresis,[15]isoelectric focusing,[16] or high-performance liquid chromatography.[17] Diagnosis is commonly confirmed by DNA sequencing.[17]
The hemoglobin structural variants can be broadly classified as follows:[18]
Sickle cell disorders, which are the most prevalent form of hemoglobinopathy. Sickle hemoglobin (HbS) is prone to polymerize when deoxygenated, precipitating within the red blood cell. This damages the RBC membrane resulting in its premature destruction and consequent anemia.[19]
Unstable hemoglobin variants are mutations that cause the hemoglobin molecule to precipitate, spontaneously or upon oxidative stress, resulting in hemolytic anemia. Precipitated, denatured hemoglobin can attach to the inner layer of the plasma membrane of the red blood cell (RBC) forming Heinz bodies, leading to premature destruction of the RBC and anemia.[20]
Change in oxygen affinity. High or low oxygen affinity hemoglobin molecules are more likely than normal to adopt the relaxed (R, oxy) state or the tense (T, deoxy) state, respectively. High oxygen affinity variants (R state) cause polycythemia (e.g., Hb Chesapeake, Hb Montefiore). Low oxygen affinity variants can cause cyanosis (e.g., Hb Kansas, Hb Beth Israel).[21]
Chemical abnormalities
Methemoglobinemia is a condition caused by elevated levels of methemoglobin in the blood. Methaemoglobin is a form of hemoglobin that contains the ferric [Fe3+] form of iron, instead of the ferrous [Fe2+] form . Methemoglobin cannot bind oxygen, which means it cannot carry oxygen to tissues. In human blood a trace amount of methemoglobin is normally produced spontaneously; the enzyme methemoglobin reductase is responsible for converting methemoglobin back to hemoglobin.[22][23] Methemoglobinemia can be hereditary but more commonly occurs as a side effect of certain medications or by abuse of recreational drugs.[24]
B) Quantitative
Production abnormalities
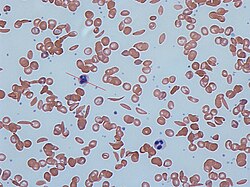
Red blood cells from a person with beta thalassemia
Thalassemias are quantitative defects that lead to reduced levels of one type of globin chain, creating an imbalance in the ratio of alpha-like chains to beta-like chains. This ratio is normally tightly regulated to prevent excess globin chains of one type from accumulating. The excess chains that fail to incorporate into normal hemoglobin can form non-functional aggregates that precipitate. This can lead to premature RBC destruction in the bone marrow and/or in the peripheral blood. Thalassemia subtypes of clinical significance are alpha thalassemia and beta thalassemia. A third subtype, delta thalassemia, affects production of HBA2 and is generally asymptomatic.[25]
The severity of alpha thalassemia depends on how many of the four genes that code for alpha globin are faulty. In the fetus, a deficiency of alpha globin results in the production of Hemoglobin Barts - a dysfunctional hemoglobin that consists of four gamma globins. In this situation, a fetus will develop hydrops fetalis and normally die before or shortly after birth.[26] In adults alpha thalassemia manifests as HbH disease. In this, excess beta-globin forms β4-tetramers, which accumulate and precipitate in red blood cells, damaging their membranes. Damaged RBCs are removed by the spleen resulting in moderate to severe anemia.[27]
In beta thalassemia, reduced production of beta globin, combined with a normal synthesis of alpha globin, results in an accumulation of excess unmatched alpha globin. This precipitates in the red cell precursors in the bone marrow, triggering their premature destruction. Anemia in beta thalassemia results from a combination of ineffective production of RBCs, peripheral hemolysis, and an overall reduction in hemoglobin synthesis.[28]
Combination hemoglobinopathies
A combination hemoglobinopathy occurs when someone inherits two different abnormal hemoglobin genes. If these are different versions of the same gene, one having been inherited from each parent it is an example of compound heterozygosity.
Both alpha- and beta- thalassemia can coexist with other hemoglobinopathies. Combinations involving alpha thalassemia are generally benign.[29][30]
Some examples of clinically significant combinations involving beta thalassemia include:
Hemoglobin C/ beta thalassemia: common in Mediterranean and African populations generally results in a moderate form of anemia with splenomegaly.[31]
Delta-beta thalassemia is a rare form of thalassemia in which there is a reduced production of both the delta and beta globins. It is generally asymptomatic.[35]
There are two clinically significant combinations involving the sickle cell gene:
Hemoglobin S/ hemoglobin C (Hemoglobin SC disease) occurs when an individual inherits one gene for hemoglobin S (sickle cell) and one gene for hemoglobin C, The symptoms are very similar to sickle cell disease.[36]
Hemoglobin variants
Hemoglobin variants are not necessarily pathological. For example, Hb Lepore-Boston and G-Waimanalo are two variants which are non-pathological.[37] There are in excess of 1,000 known hemoglobin variants.[38] A research database of hemoglobin variants is maintained by Penn State University.[39] A few of these variants are listed below.
Some hemoglobinopathies seem to have given an evolutionary benefit, especially to heterozygotes, in areas where malaria is endemic. Malaria parasites infect red blood cells, but subtly disturb normal cellular function and subvert the immune response. A number of mechanisms have been proposed to explain the increased chance of survival for the carrier of an abnormal hemoglobin trait.[41]
↑Maton, Anthea; Jean Hopkins; Charles William McLaughlin; Susan Johnson; Maryanna Quon Warner; David LaHart; Jill D. Wright (1993). Human Biology and Health. Englewood Cliffs, New Jersey, US: Prentice Hall. ISBN978-0-13-981176-0.
↑Epstein, F. H.; Hsia, C. C. W. (1998). "Respiratory Function of Hemoglobin". New England Journal of Medicine. 338 (4): 239–47. doi:10.1056/NEJM199801223380407. PMID9435331.
↑Weatherall DJ. The New Genetics and Clinical Practice, Oxford University Press, Oxford 1991.
↑Huisman TH. The structure and function of normal and abnormal haemoglobins. In: Baillière's Clinical Haematology, Higgs DR, Weatherall DJ (Eds), W.B. Saunders, London 1993. p.1.
↑Natarajan K, Townes TM, Kutlar A. Disorders of hemoglobin structure: sickle cell anemia and related abnormalities. In: Williams Hematology, 8th ed, Kaushansky K, Lichtman MA, Beutler E, et al. (Eds), McGraw-Hill, 2010. p.ch.48.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.