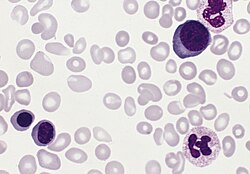
Most of the health concerns associated with PV, such as thrombosis, are caused by the blood being thicker as a result of the increased red blood cells.
PV may be asymptomatic. Possible symptoms, if any do occur, include fatigue, itching (pruritus), particularly after exposure to warm water, and severe burning pain in the hands or feet that is usually accompanied by a reddish or bluish coloration of the skin.
Treatment consists primarily of blood withdrawals (phlebotomy) and oral meds.
Approximately 98%[2][3] of PV patients have a mutation in a tyrosine kinase–encoding gene, JAK2, in their blood-forming cells[4][5] (compared with 0.1–0.2% of the general population).[6][7]
This acts in signaling pathways of the EPO receptor, making those cells proliferate independently from EPO. PV is associated with a low serum level of the hormone erythropoietin (EPO), in contrast to secondary polycythemias.[10][pageneeded]
While the mutation is a JAK2 V617F in 95% of patients, JAK2 exon 12 mutations have also been observed.[11]
Possible symptoms of PV[13][14] that may aid identification include;
pruritus (itching), particularly after exposure to warm water (such as when taking a bath),[15] which may be due to abnormal histamine release[16][17] or prostaglandin production.[18] Such itching is present in 40–55% of patients with PV.[19][20]
erythromelalgia,[21] a burning pain in the hands or feet, usually accompanied by a reddish or bluish coloration of the skin. Erythromelalgia is caused by an increased platelet count or increased platelet "stickiness" (aggregation), resulting in the formation of tiny blood clots in the vessels of the extremity; it responds rapidly to treatment with aspirin.[22][23]
Other diseases that may be present with PV include;
An enlarged spleen, a manageable condition, may occur[24] and may cause the spleen to be palpable in some patients. This may be associated with both the V617F mutation and the development of myelofibrosis.[25]
A very high red blood cell count, which is usually identified by elevated levels of hemoglobin or hematocrit;
A bone marrow biopsy that shows hypercellularity and abnormalities in megakaryocytes; and
The presence of a mutation in the Janus kinase 2 (JAK2) gene.
A minor diagnostic feature is that patients usually have a very low level of erythropoietin (EPO), a growth factor that increases the production of red blood cells.[27][11] This is used to detect cases which are negative for JAK2 mutation.[28]
Reviews 2023–25
As of 2025, reviews state diagnosis can be based on
the presence of a JAK2 mutation and
hemoglobin/hematocrit levels of >16.5 g/dL/49% in men or 16 g/dL/48% in women.
Bone marrow morphologic confirmation is advised but not mandated.[29][30]
Outlook and prognosis
Prognosis
PV may remain stable for many years, with no effect on life expectancy, particularly if managed effectively.[31] Studies show the median survival rate of controlled PV ranges from 10 to 20 years but most observations are of people diagnosed in their 60s. Patients live close to a normal life expectancy,[11] but overall survival in PV is below that of age- and sex-matched general population.[32] Factors predicting this may include age and detailed genetic differences.[32]
Possible complications and developments
PV may cause blood clotting complications (thrombosis),[33] with the two main risk factors being a previous clot or clots, and age (60 years or older).[34] If PV is untreated, there is a substantial risk of Budd-Chiari syndrome (a hepatic vein thrombosis).[35]
Additional management, depending on risks appraisal,[32][30] may include meds.[37]
A secondary treatment goal is to alleviate symptoms, for instance of pruritus (itching).[32][30]
Blood withdrawals
Blood withdrawal, sometimes called phlebotomy or venesection, is a process similar to donating blood[38] and helps to keep haematocrit levels low. This might be done weekly initially, and less often over time.[37]
Meds
Aspirin may be taken, to reduce thrombosis risk, regardless of risk category.[32]
Other medications may be used;
Hydroxyurea reduces adverse cell development. Side effects include a small increase in the risk of developing a leukaemia. Ruxolitinib (brand name Jakafi), a JAK2 inhibitor, and Busulfan may be used as alternatives.[39]
Ropeginterferon alfa-2b (Besremi) reduces the rate of blood cell production,[40][41][42] and can be used regardless of treatment history.[41] Interferon alfa-2b is also used.[43]
Anagrelide with other cytoreductive drugs may be used to manage platelet levels.[37][44]
Erlotinib may be an additional treatment option for those with certain genetic markers.[45]
Patient education and patient forums can help patients practically and emotionally manage a PV diagnosis, symptoms and other practical considerations.[36][47]
Epidemiology
Polycythemia vera occurs in all age groups,[48] although the incidence increases with age. One study found the median age at diagnosis to be 60 years,[19] and another that the highest incidence was in people aged 70–79 years.[49] 10% of PV patients are below age 40 years.[32]
Overall incidences in population studies have been 1.9/100,000 person-years in a Minnesota study,[49] and 1.48/100,000 person-years in an age-standardized Swedish study (n = 6281).[32] PV can impact all ethnic groups. There are slightly more cases in men than women.[30][49]
A cluster around a toxic site was confirmed in northeast Pennsylvania in 2008.[50]
While the JAK2 V617F mutation is generally sporadic (random), a certain inherited haplotype of JAK2 has been associated with its development.[20][51]
Figures in the discovery of and development of treatment for PV include William Osler and Louis Henri Vaquez.[56] Historically PV was called Osler–Vaquez disease.
↑Steinman H, Kobza-Black A, Lotti T, Brunetti L, Panconesi E, Greaves M (1987). "Polycythaemia rubra vera and water-induced pruritus: blood histamine levels and cutaneous fibrinolytic activity before and after water challenge". Br J Dermatol. 116 (3): 329–33. doi:10.1111/j.1365-2133.1987.tb05846.x. PMID3567071. S2CID22068469.
↑Jackson N, Burt D, Crocker J, Boughton B (1987). "Skin mast cells in polycythaemia vera: relationship to the pathogenesis and treatment of pruritus". Br J Dermatol. 116 (1): 21–9. doi:10.1111/j.1365-2133.1987.tb05787.x. PMID3814512. S2CID38261640.
↑Thurmes PJ, Steensma DP (July 2006). "Elevated serum erythropoietin levels in patients with Budd-Chiari syndrome secondary to polycythemia vera: clinical implications for the role of JAK2 mutation analysis". Eur. J. Haematol. 77 (1): 57–60. doi:10.1111/j.1600-0609.2006.00667.x. PMID16827884. S2CID37383942.
↑Passamonti F, Malabarba L, Orlandi E, Baratè C, Canevari A, Brusamolino E, Bonfichi M, Arcaini L, Caberlon S, Pascutto C, Lazzarino M (2003). "Polycythemia vera in young patients: a study on the long-term risk of thrombosis, myelofibrosis and leukemia". Haematologica. 88 (1): 13–8. PMID12551821.
123Anía B, Suman V, Sobell J, Codd M, Silverstein M, Melton L (1994). "Trends in the incidence of polycythemia vera among Olmsted County, Minnesota residents, 1935–1989". Am J Hematol. 47 (2): 89–93. doi:10.1002/ajh.2830470205. PMID8092146. S2CID31536624.
↑Berlin, N. I.; Wasserman, L. R. (Oct 25, 1997). "Polycythemia vera: a retrospective and reprise". The Journal of Laboratory and Clinical Medicine. 130 (4): 365–373. doi:10.1016/s0022-2143(97)90035-4. PMID9358074.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.