
A myelodysplastic syndrome (MDS) is one of a group of cancers in which immature blood cells in the bone marrow do not mature, and as a result, do not develop into healthy blood cells. Early on, no symptoms typically are seen. Later, symptoms may include fatigue, shortness of breath, bleeding disorders, anemia, or frequent infections. Some types may develop into acute myeloid leukemia.

Fanconi anemia (FA) is a rare, autosomal recessive, genetic disease resulting in impaired response to DNA damage in the FA/BRCA pathway. Although it is a very rare disorder, study of this and other bone marrow failure syndromes has improved scientific understanding of the mechanisms of normal bone marrow function and development of cancer. Among those affected, the majority develop cancer, most often acute myelogenous leukemia (AML), MDS, and liver tumors. 90% develop aplastic anemia by age 40. About 60–75% have congenital defects, commonly short stature, abnormalities of the skin, arms, head, eyes, kidneys, and ears, and developmental disabilities. Around 75% have some form of endocrine problem, with varying degrees of severity. 60% of FA is FANC-A, 16q24.3, which has later onset bone marrow failure.

In hematology, essential thrombocythemia (ET) is a rare chronic blood cancer characterised by the overproduction of platelets (thrombocytes) by megakaryocytes in the bone marrow. It may, albeit rarely, develop into acute myeloid leukemia or myelofibrosis. It is one of the blood cancers wherein the bone marrow produces too many white or red blood cells, or platelets.

Lenalidomide, sold under the brand name Revlimid among others, is a medication used to treat multiple myeloma, smoldering myeloma, and myelodysplastic syndromes (MDS). For multiple myeloma, it is a first line treatment, and is given with dexamethasone. It is taken by mouth.

Haploinsufficiency in genetics describes a model of dominant gene action in diploid organisms, in which a single copy of the wild-type allele at a locus in heterozygous combination with a variant allele is insufficient to produce the wild-type phenotype. Haploinsufficiency may arise from a de novo or inherited loss-of-function mutation in the variant allele, such that it yields little or no gene product. Although the other, standard allele still produces the standard amount of product, the total product is insufficient to produce the standard phenotype. This heterozygous genotype may result in a non- or sub-standard, deleterious, and (or) disease phenotype. Haploinsufficiency is the standard explanation for dominant deleterious alleles.

An isochromosome is an unbalanced structural abnormality in which the arms of the chromosome are mirror images of each other. The chromosome consists of two copies of either the long (q) arm or the short (p) arm because isochromosome formation is equivalent to a simultaneous duplication and deletion of genetic material. Consequently, there is partial trisomy of the genes present in the isochromosome and partial monosomy of the genes in the lost arm.
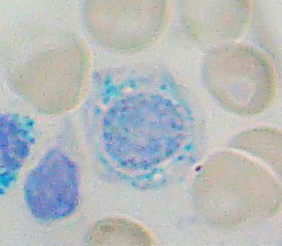
Sideroblastic anemia, or sideroachrestic anemia, is a form of anemia in which the bone marrow produces ringed sideroblasts rather than healthy red blood cells (erythrocytes). In sideroblastic anemia, the body has iron available but cannot incorporate it into hemoglobin, which red blood cells need in order to transport oxygen efficiently. The disorder may be caused either by a genetic disorder or indirectly as part of myelodysplastic syndrome, which can develop into hematological malignancies.
Diamond–Blackfan anemia (DBA) is a congenital erythroid aplasia that usually presents in infancy. DBA causes low red blood cell counts (anemia), without substantially affecting the other blood components, which are usually normal. This is in contrast to Shwachman–Bodian–Diamond syndrome, in which the bone marrow defect results primarily in neutropenia, and Fanconi anemia, where all cell lines are affected resulting in pancytopenia. There is a risk to develop acute myelogenous leukemia (AML) and certain other cancers.
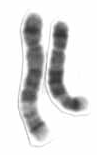
Chromosome 5 is one of the 23 pairs of chromosomes in humans. People normally have two copies of this chromosome. Chromosome 5 spans about 182 million base pairs and represents almost 6% of the total DNA in cells. Chromosome 5 is the 5th largest human chromosome, yet has one of the lowest gene densities. This is partially explained by numerous gene-poor regions that display a remarkable degree of non-coding and syntenic conservation with non-mammalian vertebrates, suggesting they are functionally constrained.
Mean platelet volume (MPV) is a machine-calculated measurement of the average size of platelets found in blood and is typically included in blood tests as part of the CBC. Since the average platelet size is larger when the body is producing increased numbers of platelets, the MPV test results can be used to make inferences about platelet production in bone marrow or platelet destruction problems.

GATA-binding factor 1 or GATA-1 is the founding member of the GATA family of transcription factors. This protein is widely expressed throughout vertebrate species. In humans and mice, it is encoded by the GATA1 and Gata1 genes, respectively. These genes are located on the X chromosome in both species.

A promyelocyte is a granulocyte precursor, developing from the myeloblast and developing into the myelocyte. Promyelocytes measure 12–20 microns in diameter. The nucleus of a promyelocyte is approximately the same size as a myeloblast but their cytoplasm is much more abundant. They also have less prominent nucleoli than myeloblasts and their chromatin is more coarse and clumped. The cytoplasm is basophilic and contains primary red/purple granules.

Chronic myelomonocytic leukemia (CMML) is a type of leukemia, which are cancers of the blood-forming cells of the bone marrow. In adults, blood cells are formed in the bone marrow, by a process that is known as haematopoiesis. In CMML, there are increased numbers of monocytes and immature blood cells (blasts) in the peripheral blood and bone marrow, as well as abnormal looking cells (dysplasia) in at least one type of blood cell.

Acute myeloblastic leukemia with maturation (M2) is a subtype of acute myeloid leukemia (AML).

Platelet-derived growth factor receptor beta is a protein that in humans is encoded by the PDGFRB gene. Mutations in PDGFRB are mainly associated with the clonal eosinophilia class of malignancies.

Acute megakaryoblastic leukemia (AMKL) is life-threatening leukemia in which malignant megakaryoblasts proliferate abnormally and injure various tissues. Megakaryoblasts are the most immature precursor cells in a platelet-forming lineage; they mature to promegakaryocytes and, ultimately, megakaryocytes which cells shed membrane-enclosed particles, i.e. platelets, into the circulation. Platelets are critical for the normal clotting of blood. While malignant megakaryoblasts usually are the predominant proliferating and tissue-damaging cells, their similarly malignant descendants, promegakaryocytes and megakaryocytes, are variable contributors to the malignancy.
In genetics, virtual karyotype is the digital information reflecting a karyotype, resulting from the analysis of short sequences of DNA from specific loci all over the genome, which are isolated and enumerated. It detects genomic copy number variations at a higher resolution for level than conventional karyotyping or chromosome-based comparative genomic hybridization (CGH). The main methods used for creating virtual karyotypes are array-comparative genomic hybridization and SNP arrays.
Bone marrow failure occurs in individuals who produce an insufficient amount of red blood cells, white blood cells or platelets. Red blood cells transport oxygen to be distributed throughout the body's tissue. White blood cells fight off infections that enter the body. Bone marrow progenitor cells known as megakaryocytes produce platelets, which trigger clotting, and thus help stop the blood flow when a wound occurs.
Ribosomopathies are diseases caused by abnormalities in the structure or function of ribosomal component proteins or rRNA genes, or other genes whose products are involved in ribosome biogenesis.

The Emberger syndrome is a rare, autosomal dominant, genetic disorder caused by familial or sporadic inactivating mutations in one of the two parental GATA2 genes. The mutation results in a haploinsufficiency in the levels of the gene's product, the GATA2 transcription factor. This transcription factor is critical for the embryonic development, maintenance, and functionality of blood-forming, lympathic-forming, and other tissues. The syndrome includes as its primary symptoms: serious abnormalities of the blood such as the myelodysplastic syndrome and acute myeloid leukemia; lymphedema of the lower limbs, and sensorineural hearing loss. However, the anomalies caused by GATA2 mutations are highly variable with some individuals showing little or no such symptoms even in old age while others exhibit non-malignant types of hematological anomalies; lymphedema in areas besides the lower limbs, little or no hearing loss; or anomalies in other tissues. The syndrome may present with relatively benign signs and/or symptoms and then progress rapidly or slowly to the myelodysplastic syndrome and/or acute myeloid leukemia. Alternatively, it may present with one of the latter two life-threatening disorders.
