p53, also known as tumor protein p53, TP53, cellular tumor antigen p53 (UniProt name), or transformation-related protein 53 (TRP53) is a regulatory transcription factor protein that is often mutated in human cancers. The p53 proteins (originally thought to be, and often spoken of as, a single protein) are crucial in vertebrates, where they prevent cancer formation.[5] As such, p53 has been described as "the guardian of the genome" because of its role in conserving stability by preventing genome mutation.[6] Hence TP53[note 1] is classified as a tumor suppressor gene.[7][8][9][10][11]
The TP53 gene is the most frequently mutated gene (>50%) in human cancer, indicating that the TP53 gene plays a crucial role in preventing cancer formation.[5]
Gene
In humans, the TP53 gene is located on the short arm of chromosome 17 (17p13.1).[7][8][9][10] The gene spans 20 kb, with a non-coding exon 1 and a very long first intron of 10 kb, overlapping the Hp53int1 gene. The coding sequence contains five regions showing a high degree of conservation in vertebrates, predominantly in exons 2, 5, 6, 7 and 8, but the sequences found in invertebrates show only distant resemblance to mammalian TP53.[12]TP53orthologs[13] have been identified in most mammals for which complete genome data are available. Elephants, with 20 genes for TP53, rarely get cancer.[14]
Structure
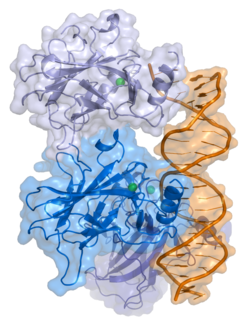
A schematic of the known protein domains in p53 (NLS = Nuclear Localization Signal)Crystal structure of four p53 DNA-binding domains (as found in the bioactive homo-tetramer)
The full-length p53 protein (p53α) comprises seven distinct protein domains:
An acidic N-terminustransactivation domain (TAD), including activation domains 1 and 2 (AD1: residues 1–42; AD2: residues 43–63), which regulate transcription of several pro-apoptotic genes.[15]
A proline-rich domain (residues 64–92), involved in apoptotic function and nuclear export via MAPK signaling.
A central DNA-binding domain (DBD; residues 102–292), containing a zinc atom and multiple arginine residues, essential for sequence-specific DNA interaction and co-repressor binding such as LMO3.[16]
A homo-oligomerization domain (OD; residues 307–355), which mediates tetramerization—essential for p53 activity in vivo.
A C-terminal regulatory domain (residues 356–393), which modulates the DNA-binding activity of the central domain.[17]
Most cancer-associated mutations in TP53 occur in the DBD, impairing DNA binding and transcriptional activation. These are typically recessive loss-of-function mutations. By contrast, mutations in the OD can exert dominant negative effects by forming inactive complexes with wild-type p53.
Although designated as a 53kDa protein by SDS-PAGE, the actual molecular weight of p53α is 43.7kDa. The discrepancy is due to its high proline content, which slows electrophoretic migration.[19]
Tetramerization
p53 initially forms dimers cotranslationally during protein synthesis on ribosomes.[20] Each dimer consists of two p53 monomers joined through their oligomerization domains.[21]
The dimerization interface spans residues 325–356 and includes a beta-strand (residues 325–333), a alpha-helix (residues 335–356), and a sharp turn at the conserved hinge residue Gly334. This configuration links the beta-strand and alpha-helix to form a V-shaped monomer topology. The beta-strand contributes to the formation of an antiparallel intermolecular beta-sheet between two p53 monomers, stabilized by hydrophobic interactions involving Phe328, Leu330, and Ile332. The alpha-helix forms an antiparallel coiled-coil between the two monomers, with a packing angle of 156°. Helix–helix interactions are stabilized by hydrophobic contacts (e.g., Phe338, Phe341, Leu344) and electrostatic interactions, such as the Arg337–Asp352 salt bridge.
Following dimer formation, p53 dimers associate posttranslationally to form tetramers (dimers of dimers).[20][22] The tetramerization domain (residues 325–356) plays a central role in stabilizing the tetrameric structure.[22] In the tetramer, the two primary dimers associate at an angle described as "roughly orthogonal," with a helix bundle packing angle (θ) of approximately 80°.
Tetramers represent the active form of p53 for DNA binding and transcriptional regulation.[23][21]
Isoforms
Like 95% of human genes, TP53 encodes multiple proteins, collectively known as the p53 isoforms.[5] These vary in size from 3.5 to 43.7kDa. Since their initial discovery in 2005, 12 human p53 isoforms have been identified: p53α, p53β, p53γ, ∆40p53α, ∆40p53β, ∆40p53γ, ∆133p53α, ∆133p53β, ∆133p53γ, ∆160p53α, ∆160p53β, and ∆160p53γ. Isoform expression is tissue-dependent, and p53α is never expressed alone.[11]
The isoforms differ by the inclusion or exclusion of specific domains. Some, such as Δ133p53β/γ and Δ160p53α/β/γ, lack the transactivation or proline-rich domains and are deficient in apoptosis induction, illustrating the functional diversity of TP53.[24][25]
Isoforms are generated through multiple mechanisms:
Alternative splicing of intron 9 creates the β and γ isoforms with altered C-termini.
An internal promoter in intron 4 produces the ∆133 and ∆160 isoforms, which lack part of the TAD and DBD.
Alternative translation initiation at codons 40 or 160 results in ∆40p53 and ∆160p53 isoforms, respectively.[11]
Function
DNA damage and repair
Activation of p53 in response to stress signals initiates its transcriptional activity, leading to the activation of cellular protective pathways
p53 functions as a transcription factor by binding DNA as a tetramer, a structure that is essential for its stability and effective DNA binding activity.[28] Once bound to DNA, p53 induces the transcription of numerous genes involved in DNA repair pathways. This includes components of base excision repair (BER) such as OGG1 and MUTYH, nucleotide excision repair (NER) factors like DDB2 and XPC, mismatch repair (MMR) genes such as MSH2 and MLH1, and elements of homologous recombination (HR) and non-homologous end-joining (NHEJ) repair.[29][30] These transcriptional responses are crucial for the DNA damage response (DDR), allowing cells to efficiently repair damaged DNA and maintain genomic integrity. While p53's role is most clearly defined in transcriptional activation of repair genes, it also participates in non-transcriptional regulation of DNA repair processes, particularly in HR and NHEJ, by modulating protein interactions and chromatin accessibility.[29][31]
p53 binds specific elements in the promoter of target genes, including CDKN1A, which encodes p21.[28][32] Upon activation by p53, p21 inhibits cyclin-dependent kinases, leading to cell cycle arrest and contributing to tumor suppression.[28][33] However, p21 can also be induced independently of p53 during processes such as differentiation, development, and in response to serum stimulation.[32]
p21 (WAF1) binds to cyclin-CDK complexes (notably CDK2, CDK1, CDK4, and CDK6), inhibiting their activity and blocking the G1/S transition.[34][35] This inhibition enforces a cell cycle pause that allows DNA repair to occur. In cells with functional p53, p21 is upregulated in response to DNA damage, ensuring this checkpoint control. In contrast, p53 mutations impair p21 induction and compromise this control.[28]
In human embryonic stem cells (hESCs), although p21 mRNA is upregulated following DNA damage, the protein is not detectable. This reflects a nonfunctional p53-p21 axis at the G1/S checkpoint.[36] This discrepancy is largely due to post-transcriptional repression, particularly by the miR-302 family of microRNAs, which inhibit p21 translation.[37] Although p53 binds the CDKN1A promoter in hESCs, it does not regulate miR-302, which is constitutively expressed and suppresses p21 expression.[37][36]
The p53 pathway is interconnected with the RB1 pathway via p14^ARF, which links the regulation of these key tumor suppressors.[38]
Levels of p53 play an important role in the maintenance of stem cells throughout development and the rest of human life.[41]
In human embryonic stem cells (hESCs)s, p53 is maintained at low inactive levels.[42] This is because activation of p53 leads to rapid differentiation of hESCs.[43] Studies have shown that knocking out p53 delays differentiation and that adding p53 causes spontaneous differentiation, showing how p53 promotes differentiation of hESCs and plays a key role in cell cycle as a differentiation regulator. When p53 becomes stabilized and activated in hESCs, it increases p21 to establish a longer G1. This typically leads to abolition of S-phase entry, which stops the cell cycle in G1, leading to differentiation. Work in mouse embryonic stem cells has recently shown however that the expression of P53 does not necessarily lead to differentiation.[44] p53 also activates miR-34a and miR-145, which then repress the hESCs pluripotency factors, further instigating differentiation.[42]
In adult stem cells, p53 regulation is important for maintenance of stemness in adult stem cell niches. Mechanical signals such as hypoxia affect levels of p53 in these niche cells through the hypoxia inducible factors, HIF-1α and HIF-2α. While HIF-1α stabilizes p53, HIF-2α suppresses it.[45] Suppression of p53 plays important roles in cancer stem cell phenotype, induced pluripotent stem cells and other stem cell roles and behaviors, such as blastema formation. Cells with decreased levels of p53 have been shown to reprogram into stem cells with a much greater efficiency than normal cells.[46][47] Papers suggest that the lack of cell cycle arrest and apoptosis gives more cells the chance to be reprogrammed. Decreased levels of p53 were also shown to be a crucial aspect of blastema formation in the legs of salamanders.[48] p53 regulation is very important in acting as a barrier between stem cells and a differentiated stem cell state, as well as a barrier between stem cells being functional and being cancerous.[49]
Other
An overview of the molecular mechanism of action of p53 on the angiogenesis
Apart from the cellular and molecular effects above, p53 has a tissue-level anticancer effect that works by inhibiting angiogenesis.[50] As tumors grow they need to recruit new blood vessels to supply them, and p53 inhibits that by (i) interfering with regulators of tumor hypoxia that also affect angiogenesis, such as HIF1 and HIF2, (ii) inhibiting the production of angiogenic promoting factors, and (iii) directly increasing the production of angiogenesis inhibitors, such as arresten.[51][52]
The immune response to infection also involves p53 and NF-κB. Checkpoint control of the cell cycle and of apoptosis by p53 is inhibited by some infections such as Mycoplasma bacteria,[54] raising the specter of oncogenic infection.
Regulation
p53 pathway: In a normal cell, p53 is inactivated by its negative regulator, mdm2. Upon DNA damage or other stress, the p53-mdm2 complex dissociates. Activated p53 can induce cell cycle arrest for repair or initiate apoptosis. The mechanism behind this decision is not fully understood.
Basal regulation
Under normal, unstressed conditions, p53 is maintained at low levels through continuous degradation mediated by the E3 ubiquitin ligaseMDM2 (HDM2 in humans).[55] MDM2 binds p53, exports it from the nucleus, and targets it for proteasomal degradation. Notably, p53 transcriptionally activates MDM2, establishing a classic negative feedback loop.
This feedback loop gives rise to damped oscillations in p53 levels, as demonstrated both experimentally[56] and in mathematical models.[57][58] These oscillations may determine cell fate decisions between survival and apoptosis.[59]
Activation involves stabilization of the p53 protein, resulting in its accumulation in the nucleus, and regulatory changes that promote sequence-specific DNA binding and transcriptional activation of target genes.[61][62] These processes are initiated in part by phosphorylation of residues in the N-terminal transactivation domain by stress-activated kinases.[61][62] Phosphorylation of sites within the Mdm2-binding region (for example Ser20) can reduce binding to MDM2 and thereby decrease ubiquitin-mediated degradation of p53.[63][64]
Stress-responsive kinases
Kinases that regulate p53 phosphorylation can be divided into two broad groups. One group includes members of the MAPK pathways, including JNK1–3, ERK1/2 and p38 MAPK, which are activated by diverse cellular stresses such as oxidative stress and heat shock.[65] A second group comprises DNA damage response kinases, including ATM, ATR and DNA-PK, together with downstream checkpoint kinases such as CHK1 and CHK2, which are activated by DNA damage and replication stress and contribute to p53 regulation through phosphorylation-dependent signalling.[66][67]
Additional kinases implicated in p53 phosphorylation include the CDK-activating kinase (CAK; CDK7–cyclin H–MAT1), which has been shown to phosphorylate p53 (for example at Ser33) in vitro and in vivo,[68] and TP53RK (PRPK), which has been reported to phosphorylate p53 at Ser15.[69]
Oncogene-induced activation of p53 can also occur via p14ARF (ARF), which inhibits the p53 antagonist MDM2 and thereby stabilizes p53.[70][71]
Deubiquitination
Several deubiquitinating enzymes (DUBs) modulate p53 stability by removing ubiquitin chains. USP7, also known as HAUSP, can deubiquitinate both p53 and MDM2. In unstressed cells, HAUSP preferentially stabilizes MDM2, and its depletion may paradoxically increase p53 levels. USP42 is another DUB that stabilizes p53 and enhances its ability to respond to stress.[72]USP10 operates primarily in the cytoplasm, where it counteracts MDM2 by directly deubiquitinating p53. After DNA damage, USP10 translocates to the nucleus and further stabilizes p53. It does not interact with MDM2.[73]
Post-translational modifications and cofactors
Phosphorylation of the N-terminus not only prevents MDM2 binding but also facilitates the recruitment of cofactors. Pin1 enhances conformational changes in p53, while p300 and PCAF acetylate the C-terminus, exposing the DNA-binding domain and enhancing transcriptional activation. Conversely, deacetylases such as Sirt1 and Sirt7 remove these modifications, suppressing apoptosis and promoting cell survival.[74] Some oncogenes can also activate p53 indirectly by inhibiting MDM2.[75]
Dynamics
Both experimental evidence and mathematical modeling indicate that p53 levels oscillate over time in response to cellular signals. These oscillations become more pronounced in the presence of DNA damage, such as double-stranded breaks or UV exposure. Modeling approaches also help illustrate how mutations in p53 isoforms affect oscillatory behavior, potentially informing tissue-specific therapeutic development.[76][77][57]
Epigenetics
p53 function is also influenced by chromatin environment. The corepressor TRIM24 restricts p53 binding to epigenetically repressed loci by recognizing methylated histones. This interaction enables p53 to interpret local chromatin context and regulate gene expression in a locus-specific manner.[78][citation needed]
Role in disease
Overview of signal transduction pathways involved in apoptosisA micrograph showing cells with abnormal p53 expression (brown) in a brain tumor. p53 immunostain.
If the TP53 gene is damaged, its ability to suppress tumors is severely compromised. Individuals who inherit only one functional copy of TP53 are predisposed to developing tumors in early adulthood, a condition known as Li–Fraumeni syndrome.[citation needed]
The TP53 gene can also be altered by mutagens—such as chemicals, radiation, or certain viruses—thereby increasing the likelihood of uncontrolled cell division. More than 50 percent of human tumors harbor a mutation or deletion of the TP53 gene.[79] Loss of p53 function leads to genomic instability, frequently resulting in an aneuploidy phenotype.[80]
Certain pathogens can also disrupt p53 activity. For example, human papillomavirus (HPV) produces the viral protein E6, which binds to and inactivates p53. In conjunction with the HPV protein E7, which inactivates the cell cycle regulator pRb, this promotes repeated cell division, clinically presenting as warts. High-risk HPV types, particularly types 16 and 18, can drive the progression from benign warts to low- or high-grade cervical dysplasia, reversible precancerous lesions. Persistent cervical infection can lead to irreversible changes, including carcinoma in situ and invasive cervical cancer. These outcomes are primarily driven by viral integration into the host genome and the continued expression of the E6 and E7 oncoproteins.[81]
Mutations
Most p53 mutations are detected by DNA sequencing. However, it is known that single missense mutations can have a large spectrum from rather mild to very severe functional effects.[77]
Pathogenic mechanisms associated with p53 mutations: (A) Wild-type p53 forms homotetramers that activate gene expression. (B) Dominant-negative mutants form heterotetramers with wild-type p53, impairing transcription in heterozygous states (p53mut/+). (C) Loss-of-function arises from complete inactivation of wild-type alleles and inactivity of the mutant protein. (D) Gain-of-function mutations confer neomorphic activities, such as hijacking other transcription factors, promoting tumorigenesis. Abbreviation: WT, wild type.
The large spectrum of cancer phenotypes due to mutations in the TP53 gene is also supported by the fact that different isoforms of p53 proteins have different cellular mechanisms for prevention against cancer. Mutations in TP53 can give rise to different isoforms, preventing their overall functionality in different cellular mechanisms and thereby extending the cancer phenotype from mild to severe. Recent studies show that p53 isoforms are differentially expressed in different human tissues, and the loss-of-function or gain-of-function mutations within the isoforms can cause tissue-specific cancer or provide cancer stem cellpotential in different tissues.[11][25][83][84] TP53 mutation also hits energy metabolism and increases glycolysis in breast cancer cells.[85]
A common human polymorphism in TP53 involves a substitution of arginine for proline at codon 72 of exon 4. Numerous studies have explored the relationship between this variation and cancer susceptibility, yielding mixed results. For instance, a 2009 meta-analysis found no association between the codon 72 polymorphism and cervical cancer risk.[86]
Other studies have identified possible associations between the codon 72 polymorphism and various cancers. A 2011 study reported that the proline variant significantly increased pancreatic cancer risk in males.[87] Another study found that proline homozygosity was associated with decreased breast cancer risk in Arab women.[88] Additional research suggested that TP53 codon 72 polymorphisms, in combination with MDM2 SNP309 and A2164G, may affect susceptibility and age of onset for non-oropharyngeal cancers in women.[89] A separate 2011 study linked the polymorphism to an increased risk of lung cancer in a Korean population.[90]
However, meta-analyses published in 2011 found no significant associations between the codon 72 variant and risks of either colorectal[91] or endometrial cancer.[92] A study of a Brazilian birth cohort found an association between the arginine variant and individuals without a family history of cancer.[93] Meanwhile, another study reported that individuals with the homozygous Pro/Pro genotype had a significantly increased risk of renal cell carcinoma.[94]
Therapeutic reactivation and gene therapy
While increasing p53 levels might appear beneficial for treating cancer, sustained p53 activation can cause premature aging.[95] A more promising approach involves restoring normal, endogenous p53 function. In some tumor types, this leads to regression via apoptosis or normalization of cell growth.[96][97]
The small-molecule inhibitor MI-63 can bind to MDM2, blocking its interaction with p53 and reactivating p53 in cancers where its function is suppressed.[99]
Diagnostic and prognostic significance
This image shows different patterns of p53 expression in endometrial cancers on chromogenic immunohistochemistry, whereof all except wild-type are variably termed abnormal/aberrant/mutation-type and are strongly predictive of an underlying TP53 mutation:[100]
Wild-type, upper left: Endometrial endometrioid carcinoma showing normal wild-type pattern of p53 expression with variable proportion of tumor cell nuclei staining with variable intensity. Note, this wild-type pattern should not be reported as "positive," because this is ambiguous reporting language.
Overexpression, upper right: Endometrial endometrioid carcinoma, grade 3, with overexpression, showing strong staining in virtually all tumor cell nuclei, much stronger compared with the internal control of fibroblasts in the center. Note, there is some cytoplasmic background indicating that this staining is quite strong but this should not be interpreted as abnormal cytoplasmic pattern.
Complete absence, lower left: Endometrial serous carcinoma showing complete absence of p53 expression with internal control showing moderate to strong but variable staining. Note, wild-type pattern in normal atrophic glands at 12 and 6 o'clock.
Both cytoplasmic and nuclear, lower right: Endometrial endometrioid carcinoma showing cytoplasmic p53 expression with internal control (stroma and normal endometrial glands) showing nuclear wild-type pattern. The cytoplasmic pattern is accompanied by nuclear staining of similar intensity.
Warren Maltzman, of the Waksman Institute of Rutgers University first demonstrated that TP53 was responsive to DNA damage in the form of ultraviolet radiation.[111] In a series of publications in 1991–92, Michael Kastan of Johns Hopkins University, reported that TP53 was a critical part of a signal transduction pathway that helped cells respond to DNA damage.[112]
In 1993, p53 was voted molecule of the year by Science magazine.[113]
12Larsen S, Yokochi T, Isogai E, Nakamura Y, Ozaki T, Nakagawara A (February 2010). "LMO3 interacts with p53 and inhibits its transcriptional activity". Biochemical and Biophysical Research Communications. 392 (3): 252–7. Bibcode:2010BBRC..392..252L. doi:10.1016/j.bbrc.2009.12.010. PMID19995558.
↑Bell S, Klein C, Müller L, Hansen S, Buchner J (October 2002). "p53 contains large unstructured regions in its native state". Journal of Molecular Biology. 322 (5): 917–27. doi:10.1016/S0022-2836(02)00848-3. PMID12367518.
↑Adimoolam S, Ford JM (September 2003). "p53 and regulation of DNA damage recognition during nucleotide excision repair". DNA Repair. 2 (9): 947–54. doi:10.1016/s1568-7864(03)00087-9. PMID12967652.
↑Gatz SA, Wiesmüller L (June 2006). "p53 in recombination and repair". Cell Death and Differentiation. 13 (6): 1003–16. doi:10.1038/sj.cdd.4401903. PMID16543940.
↑Karimian A, Ahmadi Y, Yousefi B (June 2016). "Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage". DNA Repair. 42: 63–71. doi:10.1016/j.dnarep.2016.04.008. PMID27156098.
12Babaei G, Aliarab A, Asghari Vostakolaei M, Hotelchi M, Neisari R, Gholizadeh-Ghaleh Aziz S, etal. (November 2021). "Crosslink between p53 and metastasis: focus on epithelial-mesenchymal transition, cancer stem cell, angiogenesis, autophagy, and anoikis". Molecular Biology Reports. 48 (11): 7545–7557. doi:10.1007/s11033-021-06706-1. PMID34519942. S2CID237506513.
↑Teodoro JG, Evans SK, Green MR (November 2007). "Inhibition of tumor angiogenesis by p53: a new role for the guardian of the genome". Journal of Molecular Medicine (Review). 85 (11): 1175–1186. doi:10.1007/s00109-007-0221-2. PMID17589818. S2CID10094554.
↑Borchsenius SN, Daks A, Fedorova O, Chernova O, Barlev NA (January 2018). "Effects of mycoplasma infection on the host organism response via p53/NF-κB signaling". Journal of Cellular Physiology. 234 (1): 171–180. doi:10.1002/jcp.26781. PMID30146800.
↑Bykov VJ, Eriksson SE, Bianchi J, Wiman KG (February 2018). "Targeting mutant p53 for efficient cancer therapy". Nature Reviews. Cancer. 18 (2): 89–102. doi:10.1038/nrc.2017.109. PMID29242642. S2CID4552678.
↑Chong KH, Samarasinghe S, Kulasiri D (December 2013). "Mathematical modelling of p53 basal dynamics and DNA damage response". C-fACS. 259 (20th International Congress on Mathematical Modelling and Simulation): 670–6. doi:10.1016/j.mbs.2014.10.010. PMID25433195.
12Lavin MF, Gueven N (June 2006). "The complexity of p53 stabilization and activation". Cell Death and Differentiation. 13 (6): 941–950. doi:10.1038/sj.cdd.4401925. PMID16601750.
↑Blackford AN, Jackson SP (June 2017). "ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response". Molecular Cell. 66 (6): 801–817. doi:10.1016/j.molcel.2017.05.015. PMID28622525.
↑Van Maerken T, Vandesompele J, Rihani A, De Paepe A, Speleman F (December 2009). "Escape from p53-mediated tumor surveillance in neuroblastoma: switching off the p14(ARF)-MDM2-p53 axis". Cell Death and Differentiation. 16 (12): 1563–1572. doi:10.1038/cdd.2009.138. PMID19779493.
↑Llanos S, Clark PA, Rowe J, Peters G (May 2001). "Stabilization of p53 by p14ARF without relocation of MDM2 to the nucleolus". Nature Cell Biology. 3 (5): 445–452. doi:10.1038/35074506. PMID11331871.
↑Klug SJ, Ressing M, Koenig J, Abba MC, Agorastos T, Brenna SM, etal. (August 2009). "TP53 codon 72 polymorphism and cervical cancer: a pooled analysis of individual data from 49 studies". The Lancet. Oncology. 10 (8): 772–84. doi:10.1016/S1470-2045(09)70187-1. PMID19625214.
↑Sonoyama T, Sakai A, Mita Y, Yasuda Y, Kawamoto H, Yagi T, etal. (2011). "TP53 codon 72 polymorphism is associated with pancreatic cancer risk in males, smokers and drinkers". Molecular Medicine Reports. 4 (3): 489–95. doi:10.3892/mmr.2011.449. PMID21468597.
↑Alawadi S, Ghabreau L, Alsaleh M, Abdulaziz Z, Rafeek M, Akil N, etal. (September 2011). "P53 gene polymorphisms and breast cancer risk in Arab women". Medical Oncology. 28 (3): 709–15. doi:10.1007/s12032-010-9505-4. PMID20443084. S2CID207372095.
↑Piao JM, Kim HN, Song HR, Kweon SS, Choi JS, Yun WJ, etal. (September 2011). "p53 codon 72 polymorphism and the risk of lung cancer in a Korean population". Lung Cancer. 73 (3): 264–7. doi:10.1016/j.lungcan.2010.12.017. PMID21316118.
↑Wang JJ, Zheng Y, Sun L, Wang L, Yu PB, Dong JH, etal. (November 2011). "TP53 codon 72 polymorphism and colorectal cancer susceptibility: a meta-analysis". Molecular Biology Reports. 38 (8): 4847–53. doi:10.1007/s11033-010-0619-8. PMID21140221. S2CID11730631.
↑Jiang DK, Yao L, Ren WH, Wang WZ, Peng B, Yu L (December 2011). "TP53 Arg72Pro polymorphism and endometrial cancer risk: a meta-analysis". Medical Oncology. 28 (4): 1129–35. doi:10.1007/s12032-010-9597-x. PMID20552298. S2CID32990396.
↑Thurow HS, Haack R, Hartwig FP, Oliveira IO, Dellagostin OA, Gigante DP, etal. (December 2011). "TP53 gene polymorphism: importance to cancer, ethnicity and birth weight in a Brazilian cohort". Journal of Biosciences. 36 (5): 823–31. doi:10.1007/s12038-011-9147-5. PMID22116280. S2CID23027087.
↑Huang CY, Su CT, Chu JS, Huang SP, Pu YS, Yang HY, etal. (December 2011). "The polymorphisms of P53 codon 72 and MDM2 SNP309 and renal cell carcinoma risk in a low arsenic exposure area". Toxicology and Applied Pharmacology. 257 (3): 349–55. Bibcode:2011ToxAP.257..349H. doi:10.1016/j.taap.2011.09.018. PMID21982800.
↑Chumakov PM, Iotsova VS, Georgiev GP (1982). "[Isolation of a plasmid clone containing the mRNA sequence for mouse nonviral T-antigen]". Doklady Akademii Nauk SSSR (in Russian). 267 (5): 1272–5. PMID6295732.
12Kojic S, Medeot E, Guccione E, Krmac H, Zara I, Martinelli V, etal. (May 2004). "The Ankrd2 protein, a link between the sarcomere and the nucleus in skeletal muscle". Journal of Molecular Biology. 339 (2): 313–25. doi:10.1016/j.jmb.2004.03.071. PMID15136035.
↑Khanna KK, Keating KE, Kozlov S, Scott S, Gatei M, Hobson K, etal. (December 1998). "ATM associates with and phosphorylates p53: mapping the region of interaction". Nature Genetics. 20 (4): 398–400. doi:10.1038/3882. PMID9843217. S2CID23994762.
↑Westphal CH, Schmaltz C, Rowan S, Elson A, Fisher DE, Leder P (May 1997). "Genetic interactions between atm and p53 influence cellular proliferation and irradiation-induced cell cycle checkpoints". Cancer Research. 57 (9): 1664–7. PMID9135004.
↑Leu JI, Dumont P, Hafey M, Murphy ME, George DL (May 2004). "Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex". Nature Cell Biology. 6 (5): 443–50. doi:10.1038/ncb1123. PMID15077116. S2CID43063712.
↑Garkavtsev IV, Kley N, Grigorian IA, Gudkov AV (December 2001). "The Bloom syndrome protein interacts and cooperates with p53 in regulation of transcription and cell growth control". Oncogene. 20 (57): 8276–80. doi:10.1038/sj.onc.1205120. PMID11781842. S2CID13084911.
↑Abramovitch S, Werner H (2003). "Functional and physical interactions between BRCA1 and p53 in transcriptional regulation of the IGF-IR gene". Hormone and Metabolic Research. 35 (11–12): 758–62. doi:10.1055/s-2004-814154. PMID14710355. S2CID20898175.
↑Chai YL, Cui J, Shao N, Shyam E, Reddy P, Rao VN (January 1999). "The second BRCT domain of BRCA1 proteins interacts with p53 and stimulates transcription from the p21WAF1/CIP1 promoter". Oncogene. 18 (1): 263–8. doi:10.1038/sj.onc.1202323. PMID9926942. S2CID7462625.
↑Zhang H, Somasundaram K, Peng Y, Tian H, Zhang H, Bi D, etal. (April 1998). "BRCA1 physically associates with p53 and stimulates its transcriptional activity". Oncogene. 16 (13): 1713–21. doi:10.1038/sj.onc.1201932. PMID9582019. S2CID24616900.
↑Luciani MG, Hutchins JR, Zheleva D, Hupp TR (July 2000). "The C-terminal regulatory domain of p53 contains a functional docking site for cyclin A". Journal of Molecular Biology. 300 (3): 503–18. doi:10.1006/jmbi.2000.3830. PMID10884347.
↑Ababneh M, Götz C, Montenarh M (May 2001). "Downregulation of the cdc2/cyclin B protein kinase activity by binding of p53 to p34(cdc2)". Biochemical and Biophysical Research Communications. 283 (2): 507–12. doi:10.1006/bbrc.2001.4792. PMID11327730.
↑Cuddihy AR, Wong AH, Tam NW, Li S, Koromilas AE (April 1999). "The double-stranded RNA activated protein kinase PKR physically associates with the tumor suppressor p53 protein and phosphorylates human p53 on serine 392 in vitro". Oncogene. 18 (17): 2690–702. doi:10.1038/sj.onc.1202620. PMID10348343. S2CID22467088.
↑Hofmann TG, Möller A, Sirma H, Zentgraf H, Taya Y, Dröge W, etal. (January 2002). "Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2". Nature Cell Biology. 4 (1): 1–10. doi:10.1038/ncb715. PMID11740489. S2CID37789883.
↑Leung KM, Po LS, Tsang FC, Siu WY, Lau A, Ho HT, etal. (September 2002). "The candidate tumor suppressor ING1b can stabilize p53 by disrupting the regulation of p53 by MDM2". Cancer Research. 62 (17): 4890–3. PMID12208736.
↑Garkavtsev I, Grigorian IA, Ossovskaya VS, Chernov MV, Chumakov PM, Gudkov AV (January 1998). "The candidate tumour suppressor p33ING1 cooperates with p53 in cell growth control". Nature. 391 (6664): 295–8. Bibcode:1998Natur.391..295G. doi:10.1038/34675. PMID9440695. S2CID4429461.
12Shiseki M, Nagashima M, Pedeux RM, Kitahama-Shiseki M, Miura K, Okamura S, etal. (May 2003). "p29ING4 and p28ING5 bind to p53 and p300, and enhance p53 activity". Cancer Research. 63 (10): 2373–8. PMID12750254.
↑Tsai KW, Tseng HC, Lin WC (October 2008). "Two wobble-splicing events affect ING4 protein subnuclear localization and degradation". Experimental Cell Research. 314 (17): 3130–41. doi:10.1016/j.yexcr.2008.08.002. PMID18775696.
↑Frade R, Balbo M, Barel M (December 2000). "RB18A, whose gene is localized on chromosome 17q12-q21.1, regulates in vivo p53 transactivating activity". Cancer Research. 60 (23): 6585–9. PMID11118038.
↑Guo A, Salomoni P, Luo J, Shih A, Zhong S, Gu W, etal. (October 2000). "The function of PML in p53-dependent apoptosis". Nature Cell Biology. 2 (10): 730–6. doi:10.1038/35036365. PMID11025664. S2CID13480833.
↑Romanova LY, Willers H, Blagosklonny MV, Powell SN (December 2004). "The interaction of p53 with replication protein A mediates suppression of homologous recombination". Oncogene. 23 (56): 9025–33. doi:10.1038/sj.onc.1207982. PMID15489903. S2CID23482723.
↑Riva F, Zuco V, Vink AA, Supino R, Prosperi E (December 2001). "UV-induced DNA incision and proliferating cell nuclear antigen recruitment to repair sites occur independently of p53-replication protein A interaction in p53 wild type and mutant ovarian carcinoma cells". Carcinogenesis. 22 (12): 1971–8. doi:10.1093/carcin/22.12.1971. PMID11751427.
12Cowell IG, Okorokov AL, Cutts SA, Padget K, Bell M, Milner J, etal. (February 2000). "Human topoisomerase IIalpha and IIbeta interact with the C-terminal region of p53". Experimental Cell Research. 255 (1): 86–94. doi:10.1006/excr.1999.4772. PMID10666337.
↑Derbyshire DJ, Basu BP, Date T, Iwabuchi K, Doherty AJ (October 2002). "Purification, crystallization and preliminary X-ray analysis of the BRCT domains of human 53BP1 bound to the p53 tumour suppressor". Acta Crystallographica D. 58 (Pt 10 Pt 2): 1826–9. Bibcode:2002AcCrD..58.1826D. doi:10.1107/S0907444902010910. PMID12351827.
↑Sehat B, Andersson S, Girnita L, Larsson O (July 2008). "Identification of c-Cbl as a new ligase for insulin-like growth factor-I receptor with distinct roles from Mdm2 in receptor ubiquitination and endocytosis". Cancer Research. 68 (14): 5669–77. doi:10.1158/0008-5472.CAN-07-6364. PMID18632619.
↑Okamoto T, Izumi H, Imamura T, Takano H, Ise T, Uchiumi T, etal. (December 2000). "Direct interaction of p53 with the Y-box binding protein, YB-1: a mechanism for regulation of human gene expression". Oncogene. 19 (54): 6194–202. doi:10.1038/sj.onc.1204029. PMID11175333. S2CID19222684.
"p53 Knowledgebase". Lane Group at the Institute of Molecular and Cell Biology (IMCB), Singapore. Archived from the original on 2006-01-03. Retrieved 2008-04-06.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.