
The cell cycle, or cell-division cycle, is the series of events that take place in a cell that causes it to divide into two daughter cells. These events include the duplication of its DNA and some of its organelles, and subsequently the partitioning of its cytoplasm, chromosomes and other components into two daughter cells in a process called cell division.
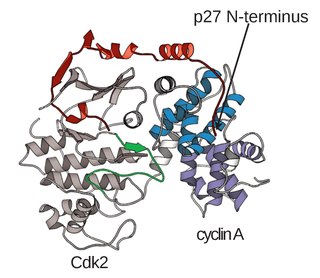
A cyclin-dependent kinase complex is a protein complex formed by the association of an inactive catalytic subunit of a protein kinase, cyclin-dependent kinase (CDK), with a regulatory subunit, cyclin. Once cyclin-dependent kinases bind to cyclin, the formed complex is in an activated state. Substrate specificity of the activated complex is mainly established by the associated cyclin within the complex. Activity of CDKCs is controlled by phosphorylation of target proteins, as well as binding of inhibitory proteins.

Cell cycle checkpoints are control mechanisms in the eukaryotic cell cycle which ensure its proper progression. Each checkpoint serves as a potential termination point along the cell cycle, during which the conditions of the cell are assessed, with progression through the various phases of the cell cycle occurring only when favorable conditions are met. There are many checkpoints in the cell cycle, but the three major ones are: the G1 checkpoint, also known as the Start or restriction checkpoint or Major Checkpoint; the G2/M checkpoint; and the metaphase-to-anaphase transition, also known as the spindle checkpoint. Progression through these checkpoints is largely determined by the activation of cyclin-dependent kinases by regulatory protein subunits called cyclins, different forms of which are produced at each stage of the cell cycle to control the specific events that occur therein.
p14ARF is an alternate reading frame protein product of the CDKN2A locus. p14ARF is induced in response to elevated mitogenic stimulation, such as aberrant growth signaling from MYC and Ras (protein). It accumulates mainly in the nucleolus where it forms stable complexes with NPM or Mdm2. These interactions allow p14ARF to act as a tumor suppressor by inhibiting ribosome biogenesis or initiating p53-dependent cell cycle arrest and apoptosis, respectively. p14ARF is an atypical protein, in terms of its transcription, its amino acid composition, and its degradation: it is transcribed in an alternate reading frame of a different protein, it is highly basic, and it is polyubiquinated at the N-terminus.

p16, is a protein that slows cell division by slowing the progression of the cell cycle from the G1 phase to the S phase, thereby acting as a tumor suppressor. It is encoded by the CDKN2A gene. A deletion in this gene can result in insufficient or non-functional p16, accelerating the cell cycle and resulting in many types of cancer.

Cyclin D is a member of the cyclin protein family that is involved in regulating cell cycle progression. The synthesis of cyclin D is initiated during G1 and drives the G1/S phase transition. Cyclin D protein is anywhere from 155 to 477 amino acids in length.

Cyclin-dependent kinase 4 also known as cell division protein kinase 4 is an enzyme that in humans is encoded by the CDK4 gene. CDK4 is a member of the cyclin-dependent kinase family.

Cell division protein kinase 6 (CDK6) is an enzyme encoded by the CDK6 gene. It is regulated by cyclins, more specifically by Cyclin D proteins and Cyclin-dependent kinase inhibitor proteins. The protein encoded by this gene is a member of the cyclin-dependent kinase, (CDK) family, which includes CDK4. CDK family members are highly similar to the gene products of Saccharomyces cerevisiae cdc28, and Schizosaccharomyces pombe cdc2, and are known to be important regulators of cell cycle progression in the point of regulation named R or restriction point.

A cyclin-dependent kinase inhibitor protein(also known as CKIs, CDIs, or CDKIs) is a protein which inhibits the enzyme cyclin-dependent kinase (CDK) and Cyclin activity by stopping the cell cycle if there are unfavorable conditions, therefore, acting as tumor suppressors. Cell cycle progression is stopped by Cyclin-dependent kinase inhibitor protein at the G1 phase. CKIs are vital proteins within the control system that point out whether the process of DNA synthesis, mitosis, and cytokines control one another. If a malfunction prevents the successful completion of DNA synthesis during the G1 phase, a signal is sent to delay or stop the progression to the S phase. Cyclin-dependent kinase inhibitor proteins are essential in the regulation of the cell cycle. If cell mutations surpass the cell cycle checkpoints during cell cycle regulation, it can result in various types of cancer.

Cyclin D1 is a protein that in humans is encoded by the CCND1 gene.

Cyclin-dependent kinase inhibitor 1B (p27Kip1) is an enzyme inhibitor that in humans is encoded by the CDKN1B gene. It encodes a protein which belongs to the Cip/Kip family of cyclin dependent kinase (Cdk) inhibitor proteins. The encoded protein binds to and prevents the activation of cyclin E-CDK2 or cyclin D-CDK4 complexes, and thus controls the cell cycle progression at G1. It is often referred to as a cell cycle inhibitor protein because its major function is to stop or slow down the cell division cycle.

G1/S-specific cyclin-D2 is a protein that in humans is encoded by the CCND2 gene.

Cyclin-dependent kinase 4 inhibitor B also known as multiple tumor suppressor 2 (MTS-2) or p15INK4b is a protein that is encoded by the CDKN2B gene in humans.
Cyclin-dependent kinase 4 inhibitor C is an enzyme that in humans is encoded by the CDKN2C gene.

Cyclin-dependent kinase 4 inhibitor D is an enzyme that in humans is encoded by the CDKN2D gene.
CDKN2A, also known as cyclin-dependent kinase inhibitor 2A, is a gene which in humans is located at chromosome 9, band p21.3. It is ubiquitously expressed in many tissues and cell types. The gene codes for two proteins, including the INK4 family member p16 and p14arf. Both act as tumor suppressors by regulating the cell cycle. p16 inhibits cyclin dependent kinases 4 and 6 and thereby activates the retinoblastoma (Rb) family of proteins, which block traversal from G1 to S-phase. p14ARF activates the p53 tumor suppressor. Somatic mutations of CDKN2A are common in the majority of human cancers, with estimates that CDKN2A is the second most commonly inactivated gene in cancer after p53. Germline mutations of CDKN2A are associated with familial melanoma, glioblastoma and pancreatic cancer. The CDKN2A gene also contains one of 27 SNPs associated with increased risk of coronary artery disease.
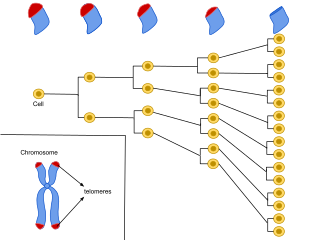
Cellular senescence is a phenomenon characterized by the cessation of cell division. In their experiments during the early 1960s, Leonard Hayflick and Paul Moorhead found that normal human fetal fibroblasts in culture reach a maximum of approximately 50 cell population doublings before becoming senescent. This process is known as "replicative senescence", or the Hayflick limit. Hayflick's discovery of mortal cells paved the path for the discovery and understanding of cellular aging molecular pathways. Cellular senescence can be initiated by a wide variety of stress inducing factors. These stress factors include both environmental and internal damaging events, abnormal cellular growth, oxidative stress, autophagy factors, among many other things.

The retinoblastoma protein is a tumor suppressor protein that is dysfunctional in several major cancers. One function of pRb is to prevent excessive cell growth by inhibiting cell cycle progression until a cell is ready to divide. When the cell is ready to divide, pRb is phosphorylated, inactivating it, and the cell cycle is allowed to progress. It is also a recruiter of several chromatin remodeling enzymes such as methylases and acetylases.
The CIP/KIP family is one of two families of mammalian cyclin dependent kinase (CDK) inhibitors (CKIs) involved in regulating the cell cycle. The CIP/KIP family is made up of three proteins: p21cip1/waf1, P27kip1, p57kip2 These proteins share sequence homology at the N-terminal domain which allows them to bind to both the cyclin and CDK. Their activity primarily involves the binding and inhibition of G1/S- and S-Cdks; however, they have also been shown to play an important role in activating the G1-CDKs CDK4 and CDK6. In addition, more recent work has shown that CIP/KIP family members have a number of CDK-independent roles involving regulation of transcription, apoptosis, and the cytoskeleton.

The Cyclin E/Cdk2 complex is a structure composed of two proteins, cyclin E and cyclin-dependent kinase 2 (Cdk2). Similar to other cyclin/Cdk complexes, the cyclin E/Cdk2 dimer plays a crucial role in regulating the cell cycle, with this specific complex peaking in activity during the G1/S transition. Once the two cyclin and Cdk subunits are joined together, the complex becomes activated and proceeds to phosphorylate and bind to downstream proteins to ultimately promote cell cycle progression. Although cyclin E can bind to other Cdk proteins, its primary binding partner is Cdk2, and the majority of cyclin E activity occurs when it exists as the cyclin E/Cdk2 complex.

