The cell cycle, or cell-division cycle, is the sequential series of events that take place in a cell that causes it to divide into two daughter cells. These events include the growth of the cell, duplication of its DNA and some of its organelles, and subsequently the partitioning of its cytoplasm, chromosomes and other components into two daughter cells in a process called cell division.

p53, also known as Tumor protein P53, cellular tumor antigen p53, or transformation-related protein 53 (TRP53) is a regulatory protein that is often mutated in human cancers. The p53 proteins are crucial in vertebrates, where they prevent cancer formation. As such, p53 has been described as "the guardian of the genome" because of its role in conserving stability by preventing genome mutation. Hence TP53 is classified as a tumor suppressor gene.

A tumor suppressor gene (TSG), or anti-oncogene, is a gene that regulates a cell during cell division and replication. If the cell grows uncontrollably, it will result in cancer. When a tumor suppressor gene is mutated, it results in a loss or reduction in its function. In combination with other genetic mutations, this could allow the cell to grow abnormally. The loss of function for these genes may be even more significant in the development of human cancers, compared to the activation of oncogenes.
p14ARF is an alternate reading frame protein product of the CDKN2A locus. p14ARF is induced in response to elevated mitogenic stimulation, such as aberrant growth signaling from MYC and Ras (protein). It accumulates mainly in the nucleolus where it forms stable complexes with NPM or Mdm2. These interactions allow p14ARF to act as a tumor suppressor by inhibiting ribosome biogenesis or initiating p53-dependent cell cycle arrest and apoptosis, respectively. p14ARF is an atypical protein, in terms of its transcription, its amino acid composition, and its degradation: it is transcribed in an alternate reading frame of a different protein, it is highly basic, and it is polyubiquinated at the N-terminus.
INK4 is a family of cyclin-dependent kinase inhibitors (CKIs). The members of this family (p16INK4a, p15INK4b, p18INK4c, p19INK4d) are inhibitors of CDK4 (hence their name INhibitors of CDK4), and of CDK6. The other family of CKIs, CIP/KIP proteins are capable of inhibiting all CDKs. Enforced expression of INK4 proteins can lead to G1 arrest by promoting redistribution of Cip/Kip proteins and blocking cyclin E-CDK2 activity. In cycling cells, there is a resassortment of Cip/Kip proteins between CDK4/5 and CDK2 as cells progress through G1. Their function, inhibiting CDK4/6, is to block progression of the cell cycle beyond the G1 restriction point. In addition, INK4 proteins play roles in cellular senescence, apoptosis and DNA repair.

Cyclin D is a member of the cyclin protein family that is involved in regulating cell cycle progression. The synthesis of cyclin D is initiated during G1 and drives the G1/S phase transition. Cyclin D protein is anywhere from 155 to 477 amino acids in length.

Cyclin-dependent kinase 4 also known as cell division protein kinase 4 is an enzyme that in humans is encoded by the CDK4 gene. CDK4 is a member of the cyclin-dependent kinase family.

Cell division protein kinase 6 (CDK6) is an enzyme encoded by the CDK6 gene. It is regulated by cyclins, more specifically by Cyclin D proteins and Cyclin-dependent kinase inhibitor proteins. The protein encoded by this gene is a member of the cyclin-dependent kinase, (CDK) family, which includes CDK4. CDK family members are highly similar to the gene products of Saccharomyces cerevisiae cdc28, and Schizosaccharomyces pombe cdc2, and are known to be important regulators of cell cycle progression in the point of regulation named R or restriction point.
The Cyclin D/Cdk4 complex is a multi-protein structure consisting of the proteins Cyclin D and cyclin-dependent kinase 4, or Cdk4, a serine-threonine kinase. This complex is one of many cyclin/cyclin-dependent kinase complexes that are the "hearts of the cell-cycle control system" and govern the cell cycle and its progression. As its name would suggest, the cyclin-dependent kinase is only active and able to phosphorylate its substrates when it is bound by the corresponding cyclin. The Cyclin D/Cdk4 complex is integral for the progression of the cell from the Growth 1 phase to the Synthesis phase of the cell cycle, for the Start or G1/S checkpoint.

Ras association domain-containing protein 1 is a protein that in humans is encoded by the RASSF1 gene.

Cyclin-dependent kinase 4 inhibitor B also known as multiple tumor suppressor 2 (MTS-2) or p15INK4b is a protein that is encoded by the CDKN2B gene in humans.
Cyclin-dependent kinase 4 inhibitor C is an enzyme that in humans is encoded by the CDKN2C gene.

Cyclin-dependent kinase 4 inhibitor D is an enzyme that in humans is encoded by the CDKN2D gene.

Hypermethylated in cancer 1 protein is a protein that in humans is encoded by the HIC1 gene.

CDKN2A, also known as cyclin-dependent kinase inhibitor 2A, is a gene which in humans is located at chromosome 9, band p21.3. It is ubiquitously expressed in many tissues and cell types. The gene codes for two proteins, including the INK4 family member p16 and p14arf. Both act as tumor suppressors by regulating the cell cycle. p16 inhibits cyclin dependent kinases 4 and 6 and thereby activates the retinoblastoma (Rb) family of proteins, which block traversal from G1 to S-phase. p14ARF activates the p53 tumor suppressor. Somatic mutations of CDKN2A are common in the majority of human cancers, with estimates that CDKN2A is the second most commonly inactivated gene in cancer after p53. Germline mutations of CDKN2A are associated with familial melanoma, glioblastoma and pancreatic cancer. The CDKN2A gene also contains one of 27 SNPs associated with increased risk of coronary artery disease.
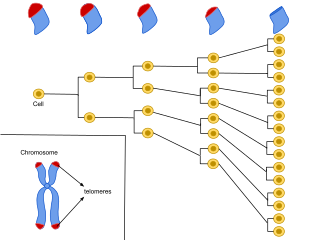
Cellular senescence is a phenomenon characterized by the cessation of cell division. In their experiments during the early 1960s, Leonard Hayflick and Paul Moorhead found that normal human fetal fibroblasts in culture reach a maximum of approximately 50 cell population doublings before becoming senescent. This process is known as "replicative senescence", or the Hayflick limit. Hayflick's discovery of mortal cells paved the path for the discovery and understanding of cellular aging molecular pathways. Cellular senescence can be initiated by a wide variety of stress inducing factors. These stress factors include both environmental and internal damaging events, abnormal cellular growth, oxidative stress, autophagy factors, among many other things.

Oropharyngeal cancer, also known as oropharyngeal squamous cell carcinoma and tonsil cancer, is a disease in which abnormal cells with the potential to both grow locally and spread to other parts of the body are found in the oral cavity, in the tissue of the part of the throat (oropharynx) that includes the base of the tongue, the tonsils, the soft palate, and the walls of the pharynx.

Human papillomavirus-positive oropharyngeal cancer, is a cancer of the throat caused by the human papillomavirus type 16 virus (HPV16). In the past, cancer of the oropharynx (throat) was associated with the use of alcohol or tobacco or both, but the majority of cases are now associated with the HPV virus, acquired by having oral contact with the genitals of a person who has a genital HPV infection. Risk factors include having a large number of sexual partners, a history of oral-genital sex or anal–oral sex, having a female partner with a history of either an abnormal Pap smear or cervical dysplasia, having chronic periodontitis, and, among men, younger age at first intercourse and a history of genital warts. HPV-positive OPC is considered a separate disease from HPV-negative oropharyngeal cancer.

Yippee-like 3 (Drosophila) is a protein that in humans is encoded by the YPEL3 gene. YPEL3 has growth inhibitory effects in normal and tumor cell lines. One of five family members (YPEL1-5), YPEL3 was named in reference to its Drosophila melanogaster orthologue. Initially discovered in a gene expression profiling assay of p53 activated MCF7 cells, induction of YPEL3 has been shown to trigger permanent growth arrest or cellular senescence in certain human normal and tumor cell types. DNA methylation of a CpG island near the YPEL3 promoter as well as histone acetylation may represent possible epigenetic mechanisms leading to decreased gene expression in human tumors.

Cancer epigenetics is the study of epigenetic modifications to the DNA of cancer cells that do not involve a change in the nucleotide sequence, but instead involve a change in the way the genetic code is expressed. Epigenetic mechanisms are necessary to maintain normal sequences of tissue specific gene expression and are crucial for normal development. They may be just as important, if not even more important, than genetic mutations in a cell's transformation to cancer. The disturbance of epigenetic processes in cancers, can lead to a loss of expression of genes that occurs about 10 times more frequently by transcription silencing than by mutations. As Vogelstein et al. points out, in a colorectal cancer there are usually about 3 to 6 driver mutations and 33 to 66 hitchhiker or passenger mutations. However, in colon tumors compared to adjacent normal-appearing colonic mucosa, there are about 600 to 800 heavily methylated CpG islands in the promoters of genes in the tumors while these CpG islands are not methylated in the adjacent mucosa. Manipulation of epigenetic alterations holds great promise for cancer prevention, detection, and therapy. In different types of cancer, a variety of epigenetic mechanisms can be perturbed, such as the silencing of tumor suppressor genes and activation of oncogenes by altered CpG island methylation patterns, histone modifications, and dysregulation of DNA binding proteins. There are several medications which have epigenetic impact, that are now used in a number of these diseases.