Steps of the cell cycle. The spindle checkpoint occurs during the M phase.Scheme showing cell cycle progression between prometaphase and anaphase.
The spindle checkpoint, also known as the metaphase-to-anaphase transition, the spindle assembly checkpoint (SAC), the metaphase checkpoint, or the mitotic checkpoint, is a cell cycle checkpoint during metaphase of mitosis or meiosis that prevents the separation of the duplicated chromosomes (anaphase) until each chromosome is properly attached to the spindle. To achieve proper segregation, the two kinetochores on the sister chromatids must be attached to opposite spindle poles (bipolar orientation).[1] Only this pattern of attachment will ensure that each daughter cell receives one copy of the chromosome. The defining biochemical feature of this checkpoint is the stimulation of the anaphase-promoting complex by M-phasecyclin-CDK complexes, which in turn causes the proteolytic destruction of cyclins and proteins that hold the sister chromatids together.[2]
The beginning of metaphase is characterized by the connection of the microtubules to the kinetochores of the chromosomes, as well as the alignment of the chromosomes in the middle of the cell. Each chromatid has its own kinetochore, and all of the microtubules that are bound to kinetochores of sister chromatids radiate from opposite poles of the cell. These microtubules exert a pulling force on the chromosomes towards the opposite ends of the cells, while the cohesion between the sister chromatids opposes this force.
At the metaphase to anaphase transition, this cohesion between sister chromatids is dissolved, and the separated chromatids are pulled to opposite sides of the cell by the spindle microtubules. The chromatids are further separated by the physical movement of the spindle poles themselves. Premature dissociation of the chromatids can lead to chromosome missegregation and aneuploidy in the daughter cells. Thus, the job of the spindle checkpoint is to prevent this transition into anaphase until the chromosomes are properly attached, before the sister chromatids separate.
In order to preserve the cell's identity and proper function, it is necessary to maintain the appropriate number of chromosomes after each cell division. An error in generating daughter cells with fewer or greater number of chromosomes than expected (a situation termed aneuploidy), may lead in best case to cell death, or alternatively it may generate catastrophic phenotypic results.[3][4] Examples include:
In cancer cells, aneuploidy is a frequent event, indicating that these cells present a defect in the machinery involved in chromosome segregation, as well as in the mechanism ensuring that segregation is correctly performed.
In humans, Down syndrome appears in children carrying in their cells one extra copy of chromosome 21, as a result of a defect in chromosome segregation during meiosis in one of the progenitors. This defect will generate a gamete (spermatozoide or oocyte) with an extra chromosome 21. After fertilisation, this gamete will generate an embryo with three copies of chromosome 21.
Discovery of the spindle assembly checkpoint (SAC)
Microscopy image showing two cells with their chromosomes stained with DAPI, one at anaphase (left) and the other in metaphase (right), with most of its chromosomes in the metaphase plate and some chromosomes still not aligned.
Zirkle (in 1970) was one of the first researchers to observe that, when just one chromosome is retarded to arrive at the metaphase plate, anaphase onset is postponed until some minutes after its arrival.[5] This observation, together with similar ones, suggested that a control mechanism exists at the metaphase-to-anaphase transition. Using drugs such as nocodazole and colchicine, the mitotic spindle disassembles and the cell cycle is blocked at the metaphase-to-anaphase transition. Using these drugs (see the review from Rieder and Palazzo in 1992[6]), the putative control mechanism was named Spindle Assembly Checkpoint (SAC). This regulatory mechanism has been intensively studied since.[7]
Using different types of genetic studies, it has been established that diverse kinds of defects are able to activate the SAC: spindle depolymerization,[8][9] the presence of dicentric chromosomes (with two centromeres),[10] centromeres segregating in an aberrant way,[11] defects in the spindle pole bodies in S. cerevisiae,[12] defects in the kinetochore proteins,[13] mutations in the centromeric DNA[14] or defects in the molecular motors active during mitosis.[8] A summary of these observations can be found in the article from Hardwick and collaborators in 1999.[15]
Using its own observations, Zirkle[5] was the first to propose that "some (…) substance, necessary for the cell to proceed to anaphase, appears some minutes after C (moment of the arrival of the last chromosome to the metaphase plate), or after a drastic change in the cytoplasmic condition, just at C or immediately after C", suggesting that this function is located on kinetochores unattached to the mitotic spindle. McIntosh extended this proposal, suggesting that one enzyme sensitive to tension located at the centromeres produces an inhibitor to the anaphase onset when the two sister kinetochores are not under bipolar tension.[16] Indeed, the available data suggested that the signal "wait to enter in anaphase" is produced mostly on or close to unattached kinetochores.[17] However, the primary event associated to the kinetochore attachment to the spindle, which is able to inactivate the inhibitory signal and release the metaphase arrest, could be either the acquisition of microtubules by the kinetochore (as proposed by Rieder and collaborators in 1995[17]), or the tension stabilizing the anchoring of microtubules to the kinetochores (as suggested by the experiments realized at Nicklas' lab[18]). Subsequent studies in cells containing two independent mitotic spindles in a sole cytoplasm showed that the inhibitor of the metaphase-to-anaphase transition is generated by unattached kinetochores and is not freely diffusible in the cytoplasm.[19] Yet in the same study it was shown that, once the transition from metaphase to anaphase is initiated in one part of the cell, this information is extended all along the cytoplasm, and can overcome the signal "wait to enter in anaphase" associated to a second spindle containing unattached kinetochores.
Background on sister chromatid duplication, cohesion, and segregation
Cell division: duplication of material and distribution to daughter cells
When cells are ready to divide, because cell size is big enough or because they receive the appropriate stimulus,[20] they activate the mechanism to enter into the cell cycle, and they duplicate most organelles during S (synthesis) phase, including their centrosome. Therefore, when the cell division process will end, each daughter cell will receive a complete set of organelles. At the same time, during S phase all cells must duplicate their DNA very precisely, a process termed DNA replication. Once DNA replication has finished, in eukaryotes the DNA molecule is compacted and condensed, to form the mitotic chromosomes, each one constituted by two sister chromatids, which stay held together by the establishment of cohesin between them; each chromatid is a complete DNA molecule, attached via microtubules to one of the two centrosomes of the dividing cell, located at opposed poles of the cell. The structure formed by the centrosomes and the microtubules is named mitotic spindle, due to its characteristic shape, holding the chromosomes between the two centrosomes. The sister chromatids stay together until anaphase, when each travels toward the centrosome to which it is attached. In this way, when the two daughter cells separate at the end of the division process, each one will contain a complete set of chromatids. The mechanism responsible for the correct distribution of sister chromatids during cell division is named chromosome segregation.
To ensure that chromosome segregation takes place correctly, cells have developed a precise and complex mechanism. In the first place, cells must coordinate centrosome duplication with DNA replication, and a failure in this coordination will generate monopolar or multipolar mitotic spindles, which generally will produce abnormal chromosome segregation,[21] because in this case, chromosome distribution will not take place in a balanced way.
Mitosis: anchoring of chromosomes to the spindle and chromosome segregation
Image of a human cell during mitosis; microtubules are shown in green (forming the mitotic spindle), chromosomes are in blue in the spindle equator and kinetochores in red.
During S phase, the centrosome starts to duplicate. Just at the beginning of mitosis, both centrioles achieve their maximal length, recruit additional material and their capacity to nucleate microtubules increases. As mitosis progresses, both centrosomes separate to generate the mitotic spindle.[22] In this way, the mitotic spindle has two poles emanating microtubules. Microtubules (MTs) are long proteic filaments, with asymmetric extremities: one end termed "minus" (-) end, relatively stable and close to the centrosome, and an end termed "plus" (+) end, with alternating phases of growth and retraction, exploring the center of the cell searching the chromosomes. Each chromatid has a special region, named the centromere, on top of which is assembled a proteic structure termed kinetochore, which is able to stabilize the microtubule plus end. Therefore, if by chance a microtubule exploring the center of the cell encounters a kinetochore, it may happen that the kinetochore will capture it, so that the chromosome will become attached to the spindle via the kinetochore of one of its sister chromatids. The chromosome plays an active role in the attachment of kinetochores to the spindle. Bound to the chromatin is a Ran guanine nucleotide exchange factor (GEF) that stimulates cytosolic Ran near the chromosome to bind GTP in place of GDP. The activated GTP-bound form of Ran releases microtubule-stabilizing proteins, such as TPX2, from protein complexes in the cytosol, which induces nucleation and polymerization of microtubules around the chromosomes.[23] These kinetochore-derived microtubules, along with kinesin motor proteins in the outer kinetochore, facilitate interactions with the lateral surface of a spindle pole-derived microtubule. These lateral attachments are unstable, however, and must be converted to an end-on attachment. Conversion from lateral to end-on attachments allows the growth and shrinkage of the microtubule plus-ends to be converted into forces that push and pull chromosomes to achieve proper bi-orientation. As it happens that sister chromatids are attached together and both kinetochores are located back-to-back on both chromatids, when one kinetochore becomes attached to one centrosome, the sister kinetochore becomes exposed to the centrosome located in the opposed pole; for this reason, in most cases the second kinetochore becomes associated to the centrosome in the opposed pole, via its microtubules,[24] so that the chromosomes become "bi-oriented", a fundamental configuration (also named amphitelic) to ensure that chromosome segregation will take place correctly when the cell will divide.[25][26] Occasionally, one of the two sister kinetochores may attach simultaneously to MTs generated by both poles, a configuration named merotelic, which is not detected by the spindle checkpoint but that may generate lagging chromosomes during anaphase and, consequently, aneuploidy. Merotelic orientation (characterized by the absence of tension between sister kinetochores) is frequent at the beginning of mitosis, but the protein Aurora B (a kinase conserved from yeast to vertebrates) detects and eliminates this type of anchoring.[27] (Aurora B is frequently overexpressed in various types of tumors and currently is a target for the development of anticancer drugs.[28])
Sister chromatid cohesion during mitosis
Cohesin: SMC proteins
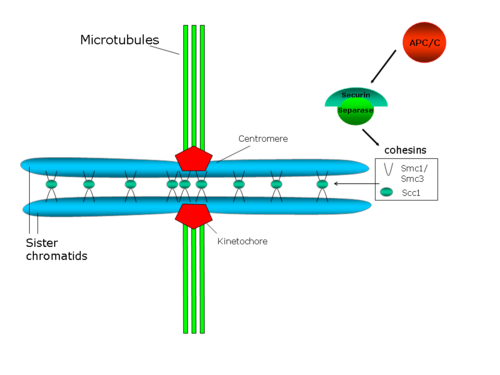
Sister chromatids stay associated from S phase (when DNA is replicated to generate two identical copies, the two chromatids) until anaphase. At this point, the two sister chromatids separate and travel to opposite poles in the dividing cell. Genetic and biochemical studies in yeast and in egg's extracts in Xenopus laevis identified a polyprotein complex as an essential player in sister chromatids cohesion (see the review from Hirano in 2000[29]). This complex is known as the cohesin complex and in Saccharomyces cerevisiae is composed of at least four subunits: Smc1p, Smc3p, Scc1p (or Mcd1p) and Scc3p. Both Smc1p and Smc3p belong to the family of proteins for the Structural Maintenance of Chromosomes (SMC), which constitute a group of chromosomic ATPases highly conserved, and form an heterodimer (Smc1p/Smc3p). Scc1p is the homolog in S.cerevisiae of Rad21, first identified as a protein involved in DNA repair in S. pombe. These four proteins are essential in yeast, and a mutation in any of them will produce premature sister chromatid separation. In yeast, cohesin binds to preferential sites along chromosome arms, and is very abundant close to the centromeres, as it was shown in a study using chromatin immunoprecipitation.[30]
The role of heterochromatin
Classical cytologic observations suggested that sister chromatids are more strongly attached at heterochromatic regions,[31] and this suggested that the special structure or composition of heterochromatin might favour cohesin recruitment.[32] In fact, it has been shown that Swi6 (the homolog of HP-1 in S. pombe) binds to methylated Lys 9 of histone H3 and promotes the binding of cohesin to the centromeric repeats in S. pombe.[33][34] More recent studies indicate that the RNAi machinery regulates heterochromatin establishment, which in turn recruits cohesin to this region, both in S. pombe[35] and in vertebrate cells.[36] However, there must be other mechanisms than heterochromatin to ensure an augmented cohesion at centromeres, because S. cerevisiae lacks heterochromatin next to centromeres, but the presence of a functional centromere induces an increase of cohesin association in a contiguous region, spanning 20-50kb.[37]
In this direction, Orc2 (one protein included in the origin recognition complex, ORC, implicated in the initiation of DNA replication during S phase) is also located on kinetochores during mitosis in human cells;[38] in agreement with this localization, some observations indicate that Orc2 in yeast is implicated in sister chromatid cohesion, and its removal induces SAC activation.[39] It has also been observed that other components of the ORC complex (such as orc5 in S. pombe) are implicated in cohesion.[40] However, the molecular pathway involving the ORC proteins seems to be additive to the cohesins' pathway, and it is mostly unknown.
Function of cohesion and its dissolution
Scheme showing sister chromatids cohesion, anchored to spindle microtubules via their kinetochores
Centromeric cohesion resists the forces exerted by spindle microtubules towards the poles, which generate tension between sister kinetochores. In turn, this tension stabilizes the attachment microtubule-kinetochore, through a mechanism implicating the protein Aurora B (a review about this issue: Hauf and Watanabe 2004[41]).
Indeed, a decrease in the cellular levels of cohesin generates the premature separation of sister chromatids, as well as defects in chromosome congression at the metaphase plate and delocalization of the proteins in the chromosomal passenger complex, which contains the protein Aurora B.[42][43] The proposed structure for the cohesin complex suggests that this complex connects directly both sister chromatids.[44] In this proposed structure, the SMC components of cohesin play a structural role, so that the SMC heterodimer may function as a DNA binding protein, whose conformation is regulated by ATP.[45] Scc1p and Scc3p, however, would play a regulatory role.[29]
In S. cerevisiae, Pds1p (also known as securin) regulates sister chromatids cohesion, because it binds and inhibits the protease Esp1p (separin or separase). When anaphase onset is triggered, the anaphase-promoting complex (APC/C or Cyclosome) degrades securin. APC/C is a ring E3 ubiquitin ligase that recruits an E2 ubiquitin-conjugating enzyme loaded with ubiquitin. Securin is recognized only if Cdc20, the activator subunit, is bound to the APC/C core. When securin, Cdc20, and E2 are all bound to APC/C E2 ubiquitinates securin and selectively degrades it. Securin degradation releases the protease Esp1p/separase, which degrades the cohesin rings that link the two sister chromatids, therefore promoting sister chromatids separation.[46] It has been also shown that Polo/Cdc5 kinase phosphorylates serine residues next to the cutting site for Scc1, and this phosphorylation would facilitate the cutting activity.[47]
Although this machinery is conserved through evolution,[48][49] in vertebrates most cohesin molecules are released in prophase, independently of the presence of the APC/C, in a process dependent on Polo-like 1 (PLK1) and Aurora B.[50] Yet it has been shown that a small quantity of Scc1 remains associated to centromeres in human cells until metaphase, and a similar amount is cut in anaphase, when it disappears from centromeres.[51] On the other hand, some experiments show that sister chromatids cohesion in the arms is lost gradually after sister centromeres have separated, and sister chromatids move toward the opposite poles of the cell.[52][53]
According to some observations, a fraction of cohesins in the chromosomal arms and the centromeric cohesins are protected by the protein Shugoshin (Sgo1), avoiding their release during prophase.[54][55] To be able to function as protector for the centromeric cohesion, Sgo1 must be inactivated at the beginning of anaphase, as well as Pds1p. In fact, both Pds1p and Sgo1 are substrates of APC/C in vertebrates.[56]
Meiosis
In mouse oocytes, DNA damage induces meiotic prophase I arrest that is mediated by the spindle assembly checkpoint.[57] Arrested oocytes do not enter the subsequent stage, anaphase I. DNA double strand breaks, UVB and ionizing radiation induced DNA damage cause an effective block to anaphase promoting complex activity.[57] This checkpoint may help prevent oocytes with damaged DNA from progressing to become fertilizable mature eggs.[57] During prophase arrest mouse oocytes appear to use both homologous recombinational repair and non-homologous end joining to repair DNA double-strand breaks.[58]
Spindle assembly checkpoint overview
The spindle assembly checkpoint (SAC) is an active signal produced by improperly attached kinetochores, which is conserved in all eukaryotes. The SAC stops the cell cycle by negatively regulating CDC20, thereby preventing the activation of the polyubiquitynation activities of anaphase promoting complex (APC). The proteins responsible for the SAC signal compose the mitotic checkpoint complex (MCC), which includes SAC proteins, MAD2/MAD3 (mitotic arrest deficient), BUB3 (budding uninhibited by benzimidazole), and CDC20.[59] Other proteins involved in the SAC include MAD1, BUB1, MPS1, and Aurora B. For higher eukaryotes, additional regulators of the SAC include constituents of the ROD-ZW10 complex, p31comet, MAPK, CDK1-cyclin-B, NEK2, and PLK1.[60]
Checkpoint activation
The SAC monitors the interaction between improperly connected kinetochores and spindle microtubules, and is maintained until kinetochores are properly attached to the spindle. During prometaphase, CDC20 and the SAC proteins concentrate at the kinetochores before attachment to the spindle assembly. These proteins keep the SAC activated until they are removed and the correct kinetochore-microtubule attachment is made. Even a single unattached kinetochore can maintain the spindle checkpoint.[59] After attachment of microtubule plus-ends and formation of kinetochore microtubules, MAD1 and MAD2 are depleted from the kinetochore assembly. Another regulator of checkpoint activation is kinetochore tension. When sister kinetochores are properly attached to opposite spindle poles, forces in the mitotic spindle generate tension at the kinetochores. Bi-oriented sister kinetochores stabilize the kinetochore-microtubule assembly whereas weak tension has a destabilizing effect. In response to incorrect kinetochore attachments such as syntelic attachment, where both kinetochores becomes attached to one spindle pole, the weak tension generated destabilizes the incorrect attachment and allows the kinetochore to reattach correctly to the spindle body. During this process, kinetochores that are attached to the mitotic spindle but that are not under tension trigger the spindle checkpoint. Aurora-B/Ipl1 kinase of the chromosomal passenger complex functions as the tensions sensor in improper kinetochore attachments. It detects and destabilizes incorrect attachments through control of the microtubule-severing KINI kinesin MCAK, the DASH complex, and the Ndc80/Hec1 complex[61] at the microtubule-kinetochore interface.[60] The Aurora-B/Ipl1 kinase is also critical in correcting merotelic attachments, where one kinetochore is simultaneously attached to both spindle poles. Merotelic attachments generate sufficient tension and are not detected by the SAC, and without correction, may result in chromosome mis-segregation due to slow chromatid migration speed. While microtubule attachment is independently required for SAC activation, it is unclear whether tension is an independent regulator of SAC, although it is clear that differing regulatory behaviors arise with tension.
Once activated, the spindle checkpoint blocks anaphase entry by inhibiting the anaphase-promoting complex via regulation of the activity of mitotic checkpoint complex. The mechanism of inhibition of APC by the mitotic checkpoint complex is poorly understood, although it is hypothesized that the MCC binds to APC as a pseudosubstrate using the KEN-box motif in BUBR1. At the same time that mitotic checkpoint complex is being activated, the centromere protein CENP-E activates BUBR1, which also blocks anaphase.[60]
Mitotic checkpoint complex formation
The mitotic checkpoint complex is composed of BUB3 together with MAD2 and MAD3 bound to Cdc20. MAD2 and MAD3 have distinct binding sites on CDC20, and act synergistically to inhibit APC/C. The MAD3 complex is composed of BUB3, which binds to Mad3 and BUB1B through the short linear motif known as the GLEBS motif. The exact order of attachments which must take place in order to form the MCC remains unknown. It is possible that Mad2-Cdc20 form a complex at the same time as BUBR1-BUB3-Cdc20 form another complex, and these two subcomplexes are consequently combined to form the mitotic checkpoint complex.[59] In human cells, binding of BUBR1 to CDC20 requires prior binding of MAD2 to CDC20, so it is possible that the MAD2-CDC20 subcomplex acts as an initiator for MCC formation. BUBR1 depletion leads only to a mild reduction in Mad2-Cdc20 levels while Mad2 is required for the binding of BubR1-Bub3 to Cdc20. Nevertheless, BUBR1 is still required for checkpoint activation.[60]
The mechanism of formation for the MCC is unclear and there are competing theories for both kinetochore-dependent and kinetochore-independent formation. In support of the kinetochore-independent theory, MCC is detectable in S. cerevisiae cells in which core kinetocore assembly proteins have been mutated and cells in which the SAC has been deactivated, which suggests that the MCC could be assembled during mitosis without kinetochore localization. In one model, unattached prometaphase kinetochores can 'sensitize' APC to inhibition of MCC by recruiting the APC to kinetochores via a functioning SAC. Furthermore, depletions of various SAC proteins have revealed that MAD2 and BUBR1 depletions affect the timing of mitosis independently of kinetochores, while depletions of other SAC proteins result in a dysfunctional SAC without altering the duration of mitosis. Thus it is possible that the SAC functions through a two-stage timer where MAD2 and BUBR1 control the duration of mitosis in the first stage, which may be extended in the second stage if there are unattached kinetochores as well as other SAC proteins.[60] However, there are lines of evidence which are in disfavor of the kinetochore-independent assembly. MCC has yet to be found during interphase, while MCC does not form from its constituents in X. laevismeiosis II extracts without the addition of sperm of nuclei and nocodazole to prevent spindle assembly.
The leading model of MCC formation is the "MAD2-template model", which depends on the kinetochore dynamics of MAD2 to create the MCC. MAD1 localizes to unattached kinetochores while binding strongly to MAD2. The localization of MAD2 and BubR1 to the kinetochore may also be dependent on the Aurora B kinase.[62] Cells lacking Aurora B fail to arrest in metaphase even when chromosomes lack microtubule attachment.[63] Unattached kinetochores first bind to a MAD1-C-MAD2-p31comet complex and releases the p31comet through unknown mechanisms. The resulting MAD1-C-MAD2 complex recruits the open conformer of Mad2 (O-Mad2) to the kinetochores. This O-Mad2 changes its conformation to closed Mad2 (C-Mad2) and binds Mad1. This Mad1/C-Mad2 complex is responsible for the recruitment of more O-Mad2 to the kinetochores, which changes its conformation to C-Mad2 and binds Cdc20 in an auto-amplification reaction. Since MAD1 and CDC20 both contain a similar MAD2-binding motif, the empty O-MAD2 conformation changes to C-MAD2 while binding to CDC20. This positive feedback loop is negatively regulated by p31comet, which competitively binds to C-MAD2 bound to either MAD1 or CDC20 and reduces further O-MAD2 binding to C-MAD2. Further control mechanisms may also exist, considering that p31comet is not present in lower eukaryotes. The 'template model' nomenclature is thus derived from the process where MAD1-C-MAD2 acts as a template for the formation of C-MAD2-CDC20 copies. This sequestration of Cdc20 is essential for maintaining the spindle checkpoint.[59]
Checkpoint deactivation
Several mechanisms exist to deactivate the SAC after correct bi-orientation of sister chromatids. Upon microtubule-kinetochore attachment, a mechanism of stripping via a dynein-dynein motor complex transports spindle checkpoint proteins away from the kinetochores.[60] The stripped proteins, which include MAD1, MAD2, MPS1, and CENP-F, are then redistributed to the spindle poles. The stripping process is highly dependent on undamaged microtubule structure as well as dynein motility along microtubules. As well as functioning as a regulator of the C-MAD2 positive feedback loop, p31comet also may act as a deactivator of the SAC. Unattached kinetochores temporarily inactivate p31comet, but attachment reactivates the protein and inhibits MAD2 activation, possibly by inhibitory phosphorylation. Another possible mechanism of SAC inactivation results from energy-dependent dissociation of the MAD2-CDC20 complex through non-degradative ubiquitylation of CDC20. Conversely, the de-ubiquitylating enzyme protectin is required to maintain the SAC. Thus, unattached kinetochores maintain the checkpoint by continuously recreating the MAD2-CDC20 subcomplex from its components. The SAC may also be deactivated by APC activation induced proteolysis. Since the SAC is not reactivated by the loss of sister-chromatid cohesion during anaphase, the proteolysis of cyclin B and inactivation of the CDK1-cyclin-B kinase also inhibits SAC activity. Degradation of MPS1 during anaphase prevents the reactivation of SAC after removal of sister-chromatid cohesion. After checkpoint deactivation and during the normal anaphase of the cell cycle, the anaphase promoting complex is activated through decreasing MCC activity. When this happens the enzyme complex polyubiquitinates the anaphase inhibitor securin. The ubiquitination and destruction of securin at the end of metaphase releases the active protease called separase. Separase cleaves the cohesion molecules that hold the sister chromatids together to activate anaphase.[23]
New model for SAC deactivation in S. cerevisiae: the mechanical switch
A new mechanism has been suggested to explain how end-on microtubule attachment at the kinetochore is able to disrupt specific steps in SAC signaling. In an unattached kinetochore, the first step in the formation of the MCC is phosphorylation of Spc105 by the kinase Mps1. Phosphorylated Spc105 is then able to recruit the downstream signaling proteins Bub1 and 3; Mad 1,2, and 3; and Cdc20. Association with Mad1 at unattached kinetochores causes Mad2 to undergo a conformational change that converts it from an open form (O-Mad2) to a closed form (C-Mad2.) The C-Mad2 bound to Mad1 then dimerizes with a second O-Mad2 and catalyzes its closure around Cdc20. This C-Mad2 and Cdc20 complex, the MCC, leaves Mad1 and C-Mad2 at the kinetochore to form another MCC. The MCCs each sequester two Cdc20 molecules to prevent their interaction with the APC/C, thereby maintaining the SAC.[23] Mps1's phosphorylation of Spc105 is both necessary and sufficient to initiate the SAC signaling pathway, but this step can only occur in the absence of microtubule attachment to the kinetochore. Endogenous Mps1 is shown to associate with the calponin-homology (CH) domain of Ndc80, which is located in the outer kinetochore region that is distant from the chromosome. Though Mps1 is docked in the outer kinetochore, it is still able to localize within the inner kinetochore and phosphorylate Spc105 because of flexible hinge regions on Ndc80. However, the mechanical switch model proposes that end-on attachment of a microtubule to the kinetochore deactivates the SAC through two mechanisms. The presence of an attached microtubule increases the distance between the Ndc80 CH domain and Spc105. Additionally, Dam1/DASH, a large complex consisting of 160 proteins that forms a ring around the attached microtubule, acts as a barrier between the two proteins. Separation prevents interactions between Mps1 and Spc105 and thus inhibits the SAC signaling pathway.[64]
This model is not applicable to SAC regulation in higher order organisms, including animals. A main facet of the mechanical switch mechanism is that in S. cerevisiae the structure of the kinetochore only allows for attachment of one microtubule. Kinetochores in animals, on the other hand, are much more complex meshworks that contain binding sites for a multitude of microtubules.[65] Microtubule attachment at all of the kinetochore binding sites is not necessary for deactivation of the SAC and progression to anaphase. Therefore, microtubule-attached and microtubule-unattached states coexist in the animal kinetochore while the SAC is inhibited. This model does not include a barrier that would prevent Mps1 associated with an attached kinetochore from phosphorylating Spc105 in an adjacent unattached kinetochore. Furthermore, the yeast Dam1/DASH complex is not present in animal cells.
Spindle checkpoint defects and cancer
When the spindle checkpoint misfunctions, this can lead to chromosome missegregation, aneuploidy and even tumorigenesis.[60] Transformation occurs and is accelerated when maintenance of genomic integrity breaks down especially at the gross level of whole chromosomes or large portions of them. In fact, aneuploidy is the most common characteristic of human solid tumors and thus the spindle assembly checkpoint might be regarded as a possible target for anti-tumour therapy.[66] This is a much underappreciated fact since mutations in specific genes known as oncogenes or tumor suppressor are primarily thought to be behind genetic instability and tumorigenesis. Usually the various checkpoints in the cell cycle take care of genomic integrity via highly conserved redundant mechanisms that are important for maintaining cellular homeostasis and preventing tumorigenesis. Several spindle assembly checkpoint proteins act both as positive and negative regulators to ensure the proper chromosome segregation in each cell cycle preventing chromosome instability (CIN) also known as genome instability.
Cytometric analysis of malignant carcinoma displaying aneuploidy
Genomic integrity is now appreciated at several levels where some tumors display instability manifested as base substitutions, insertions, and deletions, while the majority displays gains or losses of whole chromosomes.[67]
Due to the fact that alterations in mitotic regulatory proteins can lead to aneuploidy and this is a frequent event in cancer,[68] it was initially thought that these genes could be mutated in cancerous tissues.[69]
Mutated genes in cancers
In some cancers the genes that underlie the defects resulting in transformation are well characterized. In the hematological cancers such as multiple myeloma cytogenetic abnormalities are very common due to the inherent nature of DNA breaks needed for immunoglobulin gene rearrangement. However, defects in proteins such as MAD2 that function predominantly at the SAC also are characterized in multiple myeloma.[70] Most solid tumors are also predominantly aneuploid. For colorectal cancer, BUB1 and BUBR1 and amplification of STK15 are key regulators that have been implicated in the genomic instability resulting in cancer.[71] In breast cancer, the genetic form characterized by the BRCA-1 gene exhibits greater levels of genomic instability than sporadic forms. Experiments showed that BRCA-1 null mice have decreased expression of the key spindle checkpoint protein MAD2 .[72] For other cancers, more work is warranted to identify the causes of aneuploidy.
Other genes not traditionally associated with the SAC in cancer
Clearly variations in the physiological levels of these proteins (such as Mad2 or BubR1) are associated with aneuploidy and tumorigenesis, and this has been demonstrated using animal models.[73][74] However, recent studies indicate that what seems to happen is a more complicated scenario: aneuploidy would drive a high incidence of tumorigenesis only when alterations in the levels of specific mitotic checkpoint components (either reduction or overexpression) in tissues is also inducing other defects able to predispose them to tumors.[75] That is, defects such as an increase in DNA damage, chromosomal rearrangements, and/or a decreased incidence of cell death. For some mitotic checkpoint components, it is known that they are implicated in functions outside mitosis: nuclear import (Mad1), transcriptional repression (Bub3), and cell death, DNA damage response, aging, and megakaryopoiesis for BubR1. All this supports the conclusion that increase in tumorigenesis is associated with defects other than aneuploidy alone.[75]
Cancer-associated mutations affecting known checkpoint genes like BUB1 or BUBR1 are actually rare. However, several proteins implicated in cancer have intersections to spindle assembly networks. Key tumor suppressors such as p53 also play a role in the spindle checkpoint. Absence of p53, the most commonly mutated gene in human cancer, has a major effect on cell cycle checkpoint regulators and has been shown to act at the G1 checkpoint in the past, but now appears to be important in regulating the spindle checkpoint as well.[76] Another key aspect of cancer is inhibition of cell death or apoptosis. Survivin, a member of the inhibitor of apoptosis (IAP) family, is localized in pools at microtubules of the mitotic spindle near the centrosomes and at the kinetochores of metaphase chromosomes. Not only does survivin inhibit apoptosis to promote tumorigenesis, but it has been implicated (through experimental knockout mice) as an important regulator of chromosome segregation, and late stage mitosis similar to its role in more primitive organisms.[77]
Other aspects of the spindle assembly checkpoint such as kinetochore attachment, microtubule function, and sister chromatid cohesion are likely to be defective as well to cause aneuploidy. Cancer cells have been observed to divide in multiple directions by evading the spindle assembly checkpoint resulting in multipolar mitoses.[78] The multipolar metaphase-anaphase transition occurs through an incomplete separase cycle that results in frequent nondisjunction events which amplify aneuploidy in cancer cells.
SAC cancer therapies
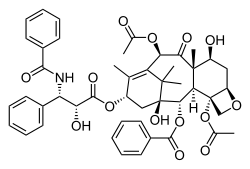
The chemical structure of paclitaxel or TAXOL, a mitotic inhibitor used in cancer chemotherapy
Advances in this field have led to the introduction of development of some therapies targeted at spindle assembly defects. Older treatments such as vinca alkaloids and taxanes target microtubules that accompany mitotic spindle formation via disruption of microtubule dynamics which engage the SAC arresting the cell and eventually leading to its death.[79]Taxol and Docetaxel, which can induce mitotic catastrophe, both are still used in the treatment of breast cancer, ovarian cancer and other types of epithelial cancer.[80] However, these treatments are often characterized by high rates of side effects and drug resistance.
Other targets within the network of regulators that influence the SAC are also being pursued; strong interest has shifted towards the aurora kinase proteins.[81] The kinase gene Aurora A when amplified acts as an oncogene overriding the SAC leading to abnormal initiation of anaphase and subsequent aneuploidy and also resistance to TAXOL .[82] Excitingly, a small molecule inhibitor of Aurora A has shown antitumor effects in an in vivo model suggesting that this might be a good target for further clinical development.[83]Aurora B inhibitors, which are also in clinical development lead to abnormal kinetochore to microtubule attachment and abrogate the mitotic checkpoint as well.[81] Survivin is also an attractive molecular target for clinical therapeutic development as it acts as a major node in a multitude of pathways, one of which is spindle formation and checkpoint control.[84] Even further approaches have included a look at inhibition of mitotic motor proteins like KSP. These inhibitors, which have recently entered clinical trials, cause mitotic arrest and by engaging the spindle assembly checkpoint and induce apoptosis.[85][3]
↑ McIntosh JR (1991). "Structural and mechanical control of mitotic progression". Cold Spring Harbor Symposia on Quantitative Biology. 56: 613–9. doi:10.1101/sqb.1991.056.01.070. PMID1819511.
↑ Gonzalez C, Casal Jimenez J, Ripoll P, Sunkel CE (January 1991). "The spindle is required for the process of sister chromatid separation in Drosophila neuroblasts". Experimental Cell Research. 192 (1): 10–5. doi:10.1016/0014-4827(91)90150-S. PMID1898588.
↑ Losada A, Hirano T (October 2001). "Shaping the metaphase chromosome: coordination of cohesion and condensation". BioEssays. 23 (10): 924–35. doi:10.1002/bies.1133. PMID11598959. S2CID31210810.
↑ Fukagawa T, Nogami M, Yoshikawa M, Ikeno M, Okazaki T, Takami Y, Nakayama T, Oshimura M (August 2004). "Dicer is essential for formation of the heterochromatin structure in vertebrate cells". Nature Cell Biology. 6 (8): 784–91. doi:10.1038/ncb1155. PMID15247924. S2CID24798145.
↑ Alberts B, Johnson A, Lewis J, Morgan D, Raff M, Roberts K, Walter P (2015). Molecular Biology of The Cell (6th ed.). New York, NY: Garland Science, Taylor & Francis Group. p.988. ISBN978-0-8153-4432-2.
↑ Kops GJ, Weaver BA, Cleveland DW (October 2005). "On the road to cancer: aneuploidy and the mitotic checkpoint". Nature Reviews. Cancer. 5 (10): 773–85. doi:10.1038/nrc1714. PMID16195750. S2CID2515388.
↑ Yamamoto Y, Matsuyama H, Chochi Y, Okuda M, Kawauchi S, Inoue R, Furuya T, Oga A, Naito K, Sasaki K (April 2007). "Overexpression of BUBR1 is associated with chromosomal instability in bladder cancer". Cancer Genetics and Cytogenetics. 174 (1): 42–7. doi:10.1016/j.cancergencyto.2006.11.012. PMID17350465.
↑ Altieri DC (December 2001). "The molecular basis and potential role of survivin in cancer diagnosis and therapy". Trends in Molecular Medicine. 7 (12): 542–7. doi:10.1016/S1471-4914(01)02243-2. PMID11733216.
↑ Harrington EA, Bebbington D, Moore J, Rasmussen RK, Ajose-Adeogun AO, Nakayama T, Graham JA, Demur C, Hercend T, Diu-Hercend A, Su M, Golec JM, Miller KM (March 2004). "VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo". Nature Medicine. 10 (3): 262–7. doi:10.1038/nm1003. PMID14981513. S2CID12918452.
↑ Altieri DC (January 2008). "Survivin, cancer networks and pathway-directed drug discovery". Nature Reviews. Cancer. 8 (1): 61–70. doi:10.1038/nrc2293. PMID18075512. S2CID25597711.
Kitagawa R, Rose AM (December 1999). "Components of the spindle-assembly checkpoint are essential in Caenorhabditis elegans". Nature Cell Biology. 1 (8): 514–21. doi:10.1038/70309. PMID10587648. S2CID25953096.
External links
Ted Salmon's lab: dividing cells movies.
Andrea Musacchio's lab: spindle checkpoint schemes.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.