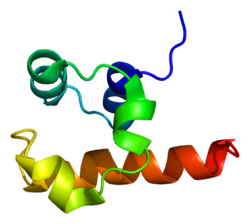
Tumor protein p63, typically referred to as p63, also known as transformation-related protein 63, is a protein that in humans is encoded by the TP63 (also known as the p63) gene.[5][6][7][8]
The TP63 gene was discovered 20 years after the discovery of the p53 tumor suppressor gene and along with p73 constitutes the p53 gene family based on their structural similarity.[9] Despite being discovered significantly later than p53, phylogenetic analysis of p53, p63 and p73, suggest that p63 was the original member of the family from which p53 and p73 evolved.[10]
Function
Tumor protein p63 is a member of the p53 family of transcription factors. p63 -/- mice have several developmental defects which include the lack of limbs and other tissues, such as teeth and mammary glands, which develop as a result of interactions between mesenchyme and epithelium. TP63 encodes for two main isoforms by alternative promoters (TAp63 and ΔNp63). ΔNp63 is involved in multiple functions during skin development and in adult stem/progenitor cell regulation.[11] In contrast, TAp63 has been mostly restricted to its apoptotic function and more recently as the guardian of oocyte integrity.[12] Recently, two new functions have been attributed to TAp63 in heart development[13] and premature aging.[14]
In mice, p63 is required for normal skin development via direct transcription of the membrane protein PERP. TP63 can also regulate PERP expression with TP53 in human cancer.[15]
Oocyte integrity
In oocytes, a unique quality control system has evolved that eliminates by apoptosis those oocytes in which chromosomes do not align correctly, or in which chromosomes cannot be repaired.[16] This monitoring system is conserved from fruit flies and nematodes to humans, and central to this system is the p53 protein family and, in vertebrates in particular, the p63 protein.[16] Oocytes that are unable to repair DNA double-strand breaks produced during meiosis by the process of homologous recombination are eliminated by apoptosis that is linked to p63.[16]
p63 staining on prostate cancer tissue using antibody clone IHC063
Both cleft lip with or without a cleft palate and cleft palate only features have been seen to segregate within the same family with a TP63 mutation.[18] Recently, induced pluripotent stem cells have been produced from patients affected by EEC syndromes by cell reprogramming. The defective epithelial commitment could be partially rescued by a small therapeutic compound.[19]
Molecular mechanism
Transcription factor p63 is a key regulator of epidermal keratinocyte proliferation and differentiation. In a recent study, researchers used EEC-patient-derived skin keratinocytes carrying heterozygous p63 DNA-binding domain mutations as the cellular model to characterize the global gene regulatory alteration. The epidermal cell identity was compromised in p63 mutant keratinocytes. Besides, p63-binding loss and loss of active enhancers occurs at a genome-wide scale in patient keratinocytes carrying heterozygous EEC mutations.[20] Besides, using a multi-omics approach, the deregulated function of DNA loops mediated by p63 and CTCF represents an additional layer to the disease mechanism. It seems that a number of loci nearby epidermal genes were organized into a 'regulatory chromatin hub' within the chromatin interactions, mediated by CTCF in epidermal keratinocytes. Such hubs contain multiple connecting DNA loops that require not only CTCF binding that is rather static but also binding of cell type-specific TFs, like p63, for the transcriptional activity. In this model, p63 may be essential to make the DNA loops active in transcription.[21]
Vulvar cancer
TP63 has been observed overexpressed in Vulvar Squamous Cell Carcinoma samples, in association with hypermethylation-Induced inactivation of the IRF6 tumor suppressor gene.[22] Indeed, mRNA levels of TP63 tested higher in Vulvar cancer samples when compared with those of normal skin and preneoplastic vulvar lesions, thus underscoring an epigenetic cross-link between IRF6 gene and the oncogene TP63.[22]
Diagnostic utility
Main staining patterns on chromogenic immunohistochemistry.
p63 immunostaining has utility for head and neck squamous cell carcinomas, differentiating prostatic adenocarcinoma (the most common type of prostate cancer) and benign prostatic tissue;[23] the nuclei of the basal cells of normal prostatic glands stain with p63, while the malignant glands in prostatic adenocarcinoma (which lacks these cells) do not.[24] P63 is also helpful in distinguishing poorly differentiated squamous cell carcinoma from small cell carcinoma or adenocarcinoma. P63 should be strongly stained in poorly differentiated squamous cell, but negative in small cell or adenocarcinoma.[25]
Cytoplasmic staining on immunohistochemistry is seen in cells with muscle differentiation.[26]
Interactions
TP63 has been shown to interact with HNRPAB.[27] It also activates IRF6 transcription through the IRF6 enhancer element.[18]
Regulation
There is some evidence that the expression of p63 is regulated by the microRNA miR-203[28][29] and USP28 at protein level [30][31]
See also
AMACR - another marker for prostate adenocarcinoma
↑Osada M, Ohba M, Kawahara C, Ishioka C, Kanamaru R, Katoh I, etal. (July 1998). "Cloning and functional analysis of human p51, which structurally and functionally resembles p53". Nature Medicine. 4 (7): 839–43. doi:10.1038/nm0798-839. PMID9662378. S2CID21953916.
↑Wu G, Nomoto S, Hoque MO, Dracheva T, Osada M, Lee CC, etal. (May 2003). "DeltaNp63alpha and TAp63alpha regulate transcription of genes with distinct biological functions in cancer and development". Cancer Research. 63 (10): 2351–7. PMID12750249.
↑Skipper M (January 2007). "Dedicated protection for the female germline". Nature Reviews Molecular Cell Biology. 8 (1): 4–5. doi:10.1038/nrm2091. S2CID10702219.
12Rotondo JC, Borghi A, Selvatici R, Magri E, Bianchini E, Montinari E, etal. (August 2016). "Hypermethylation-Induced Inactivation of the IRF6 Gene as a Possible Early Event in Progression of Vulvar Squamous Cell Carcinoma Associated With Lichen Sclerosus". JAMA Dermatology. 152 (8): 928–33. doi:10.1001/jamadermatol.2016.1336. PMID27223861.
↑Shiran MS, Tan GC, Sabariah AR, Rampal L, Phang KS (March 2007). "p63 as a complementary basal cell specific marker to high molecular weight-cytokeratin in distinguishing prostatic carcinoma from benign prostatic lesions". The Medical Journal of Malaysia. 62 (1): 36–9. PMID17682568.
↑Herawi M, Epstein JI (June 2007). "Immunohistochemical antibody cocktail staining (p63/HMWCK/AMACR) of ductal adenocarcinoma and Gleason pattern 4 cribriform and noncribriform acinar adenocarcinomas of the prostate". The American Journal of Surgical Pathology. 31 (6): 889–94. doi:10.1097/01.pas.0000213447.16526.7f. PMID17527076. S2CID9054387.
↑Prieto-Garcia C, Hartmann O, Reissland M, Braun F, Fischer T, Walz S, etal. (Jun 2019). "The USP28-∆Np63 axis is a vulnerability of squamous tumours". bioRxiv10.1101/683508.
van Bokhoven H, McKeon F (March 2002). "Mutations in the p53 homolog p63: allele-specific developmental syndromes in humans". Trends in Molecular Medicine. 8 (3): 133–9. doi:10.1016/S1471-4914(01)02260-2. PMID11879774.
Brunner HG, Hamel BC, van Bokhoven H (October 2002). "P63 gene mutations and human developmental syndromes". American Journal of Medical Genetics. 112 (3): 284–90. doi:10.1002/ajmg.10778. PMID12357472.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.