
Alternative splicing, or alternative RNA splicing, or differential splicing, is an alternative splicing process during gene expression that allows a single gene to code for multiple proteins. In this process, particular exons of a gene may be included within or excluded from the final, processed messenger RNA (mRNA) produced from that gene. This means the exons are joined in different combinations, leading to different (alternative) mRNA strands. Consequently, the proteins translated from alternatively spliced mRNAs usually contain differences in their amino acid sequence and, often, in their biological functions.
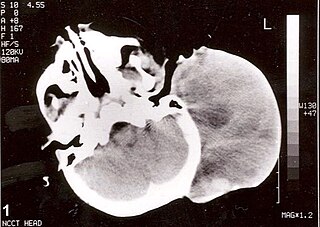
Rhabdomyosarcoma (RMS) is a highly aggressive form of cancer that develops from mesenchymal cells that have failed to fully differentiate into myocytes of skeletal muscle. Cells of the tumor are identified as rhabdomyoblasts.

L1, also known as L1CAM, is a transmembrane protein member of the L1 protein family, encoded by the L1CAM gene. This protein, of 200-220 kDa, is a neuronal cell adhesion molecule with a strong implication in cell migration, adhesion, neurite outgrowth, myelination and neuronal differentiation. It also plays a key role in treatment-resistant cancers due to its function. It was first identified in 1984 by M. Schachner who found the protein in post-mitotic mice neurons.
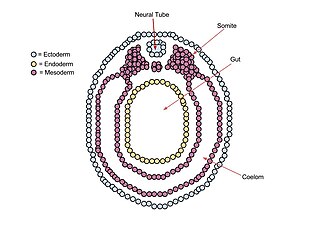
A neurula is a vertebrate embryo at the early stage of development in which neurulation occurs. The neurula stage is preceded by the gastrula stage; consequentially, neurulation is preceded by gastrulation. Neurulation marks the beginning of the process of organogenesis.

In genetics, a silencer is a DNA sequence capable of binding transcription regulation factors, called repressors. DNA contains genes and provides the template to produce messenger RNA (mRNA). That mRNA is then translated into proteins. When a repressor protein binds to the silencer region of DNA, RNA polymerase is prevented from transcribing the DNA sequence into RNA. With transcription blocked, the translation of RNA into proteins is impossible. Thus, silencers prevent genes from being expressed as proteins.

In evolutionary developmental biology, Paired box (Pax) genes are a family of genes coding for tissue specific transcription factors containing an N-terminal paired domain and usually a partial, or in the case of four family members, a complete homeodomain to the C-terminus. An octapeptide as well as a Pro-Ser-Thr-rich C terminus may also be present. Pax proteins are important in early animal development for the specification of specific tissues, as well as during epimorphic limb regeneration in animals capable of such.

Paired box protein Pax-6, also known as aniridia type II protein (AN2) or oculorhombin, is a protein that in humans is encoded by the PAX6 gene.

Myogenesis is the formation of skeletal muscular tissue, particularly during embryonic development.

Fibroblast growth factor receptor 1 (FGFR1), also known as basic fibroblast growth factor receptor 1, fms-related tyrosine kinase-2 / Pfeiffer syndrome, and CD331, is a receptor tyrosine kinase whose ligands are specific members of the fibroblast growth factor family. FGFR1 has been shown to be associated with Pfeiffer syndrome, and clonal eosinophilias.

The steroidogenic factor 1 (SF-1) protein is a transcription factor involved in sex determination by controlling the activity of genes related to the reproductive glands or gonads and adrenal glands. This protein is encoded by the NR5A1 gene, a member of the nuclear receptor subfamily, located on the long arm of chromosome 9 at position 33.3. It was originally identified as a regulator of genes encoding cytochrome P450 steroid hydroxylases, however, further roles in endocrine function have since been discovered.
Alveolar rhabdomyosarcoma (ARMS) is a subtype of the rhabdomyosarcoma soft tissue cancer family whose lineage is from mesenchymal cells and are related to skeletal muscle cells. ARMS tumors resemble the alveolar tissue in the lungs. Tumor location varies from patient to patient, but is commonly found in the head and neck region, male and female urogenital tracts, the torso, and extremities. Two fusion proteins can be associated with ARMS, but are not necessary, PAX3-FKHR. and PAX7-FKHR. In children and adolescents ARMS accounts for about 1 percent of all malignancies, has an incidence rate of 1 per million, and most cases occur sporadically with no genetic predisposition. PAX3-FOXO1 is now known to drive cancer-promoting gene expression programs through creation of distant genetic elements called super enhancers.
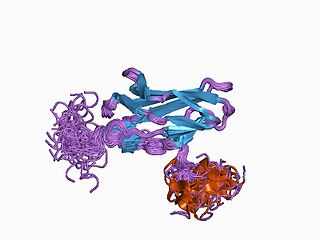
Runt-related transcription factor 1 (RUNX1) also known as acute myeloid leukemia 1 protein (AML1) or core-binding factor subunit alpha-2 (CBFA2) is a protein that in humans is encoded by the RUNX1 gene.

Protein C-ets-1 is a protein that in humans is encoded by the ETS1 gene. The protein encoded by this gene belongs to the ETS family of transcription factors.
Malignant ectomesenchymoma(MEM) is a rare, fast-growing tumor of the nervous system or soft tissue that occurs in children and young adults. MEM is part of a group of small round blue cell tumors which includes neuroblastoma, rhabdomyosarcoma, non-Hodgkin's lymphoma, and the Ewing's family of tumors.

Paired box gene 8, also known as PAX8, is a protein which in humans is encoded by the PAX8 gene.
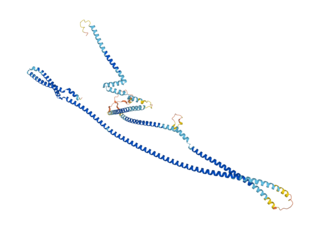
Cux1 is a homeodomain protein that in humans is encoded by the CUX1 gene.

Paired box protein Pax-7 is a protein that in humans is encoded by the PAX7 gene.

Forkhead box protein O1 (FOXO1), also known as forkhead in rhabdomyosarcoma (FKHR), is a protein that in humans is encoded by the FOXO1 gene. FOXO1 is a transcription factor that plays important roles in regulation of gluconeogenesis and glycogenolysis by insulin signaling, and is also central to the decision for a preadipocyte to commit to adipogenesis. It is primarily regulated through phosphorylation on multiple residues; its transcriptional activity is dependent on its phosphorylation state.
Embryonal rhabdomyosarcoma (EMRS) is a rare histological form of cancer in the connective tissue wherein the mesenchymally-derived malignant cells resemble the primitive developing skeletal muscle of the embryo. It is the most common soft tissue sarcoma occurring in children. Embryonal rhabdomyosarcoma is also known as PAX-fusion negative or fusion-negative rhabdomyosarcoma, as tumors of this subtype are unified by their lack of a PAX3-FOXO1 fusion oncogene. Fusion status refers to the presence or absence of a fusion gene, which is a gene formed from joining two different genes together through DNA rearrangements. These types of tumors are classified as embryonal rhabdomyosarcoma "because of their remarkable resemblance to developing embryonic and fetal skeletal muscle."

Gooseberry (gsb) is a segment polarity gene located on chromosome 2 of the Drosophila genome. Gooseberry is known for its interactions with key embryonic signaling pathways Wingless and Hedgehog. The gene also has clinal significance, being linked to diseases such as Waardenburg Syndrome and rhabdomyosarcoma.







