



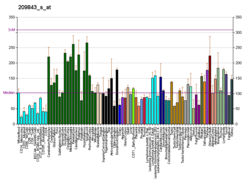

Transcription factor SOX-10 is a protein that in humans is encoded by the SOX10 gene. [5] [6] [7] [8]
Transcription factor SOX-10 is a protein that in humans is encoded by the SOX10 gene. [5] [6] [7] [8]
This gene encodes a member of the SOX (SRY-related HMG-box) family of transcription factors involved in the regulation of embryonic development and determination of cell fate. The encoded protein acts as a transcriptional activator after forming a protein complex with other proteins. This protein acts as a nucleocytoplasmic shuttle protein and is important for neural crest and peripheral nervous system development. [8]
In melanocytic cells, there is evidence that SOX10 gene expression may be regulated by MITF. [9]
Mutations in this gene are associated with Waardenburg–Shah syndrome [8] and uveal melanoma. [10]
SOX10 is used as an immunohistochemistry marker, being positive in: [11]



The interaction between SOX10 and PAX3 is studied best in human patients with Waardenburg syndrome, an autosomal dominant disorder that is divided into four different types based upon mutations in additional genes. SOX10 and PAX3 interactions are thought to be regulators of other genes involved in the symptoms of Waardenburg syndrome, particularly MITF, which influences the development of melanocytes as well as neural crest formation. MITF expression can be transactivated by both SOX10 and PAX3 to have an additive effect. [12] [13] The two genes have binding sites near one another on the upstream enhancer of the c-RET gene. [14] SOX10 is also thought to target dopachrome tautomerase through a synergistic interaction with MITF, which then results in other melanocyte alteration. [15]
SOX10 can influence the generation of Myelin Protein Zero (MPZ) transcription through its interactions with proteins such as OLIG1 and EGR2, [16] [17] which is important for the functionality of neurons. Other cofactors have been identified, such as SP1, OCT6, NMI, FOXD3 and SOX2. [18]
The interaction between SOX10 and NMI seems to be coexpressed in glial cells, gliomas, and the spinal cord and has been shown to modulate the transcriptional activity of SOX10. [19]

Waardenburg syndrome is a group of rare genetic conditions characterised by at least some degree of congenital hearing loss and pigmentation deficiencies, which can include bright blue eyes, a white forelock or patches of light skin. These basic features constitute type 2 of the condition; in type 1, there is also a wider gap between the inner corners of the eyes called telecanthus, or dystopia canthorum. In type 3, which is rare, the arms and hands are also malformed, with permanent finger contractures or fused fingers, while in type 4, the person also has Hirschsprung's disease. There also exist at least two types that can result in central nervous system (CNS) symptoms such as developmental delay and muscle tone abnormalities.

Neural crest cells are a temporary group of cells that arise from the embryonic ectoderm germ layer, and in turn give rise to a diverse cell lineage—including melanocytes, craniofacial cartilage and bone, smooth muscle, peripheral and enteric neurons and glia.
Albinism-black lock-cell migration disorder is the initialism for the following terms and concepts that describe a condition affecting a person's physical appearance and physiology: (1) A – albinism, (2) B – black lock of hair, (3) C – cell migration disorder of the neurocytes of the gut, and (4) D – sensorineural deafness. The syndrome is caused by mutation in the endothelin B receptor gene (EDNRB).

In evolutionary developmental biology, Paired box (Pax) genes are a family of genes coding for tissue specific transcription factors containing an N-terminal paired domain and usually a partial, or in the case of four family members, a complete homeodomain to the C-terminus. An octapeptide as well as a Pro-Ser-Thr-rich C terminus may also be present. Pax proteins are important in early animal development for the specification of specific tissues, as well as during epimorphic limb regeneration in animals capable of such.

The PAX3 gene encodes a member of the paired box or PAX family of transcription factors. The PAX family consists of nine human (PAX1-PAX9) and nine mouse (Pax1-Pax9) members arranged into four subfamilies. Human PAX3 and mouse Pax3 are present in a subfamily along with the highly homologous human PAX7 and mouse Pax7 genes. The human PAX3 gene is located in the 2q36.1 chromosomal region, and contains 10 exons within a 100 kb region.

Microphthalmia-associated transcription factor also known as class E basic helix-loop-helix protein 32 or bHLHe32 is a protein that in humans is encoded by the MITF gene.
Zinc finger E-box-binding homeobox 2 is a protein that in humans is encoded by the ZEB2 gene. The ZEB2 protein is a transcription factor that plays a role in the transforming growth factor β (TGFβ) signaling pathways that are essential during early fetal development.

Endothelin receptor type B, (ET-B) is a protein that in humans is encoded by the EDNRB gene.

G-protein coupled receptor 143, also known as Ocular albinism type 1 (OA1) in humans, is a conserved integral membrane protein with seven transmembrane domains and similarities with G protein-coupled receptors (GPCRs) that is expressed in the eye and epidermal melanocytes. This protein encoded by the GPR143 gene, whose variants can lead to Ocular albinism type 1.

Endothelin-3 is a protein that in humans is encoded by the EDN3 gene.

Eyes absent homolog 1 is a protein that in humans is encoded by the EYA1 gene.

Gap junction beta-3 protein (GJB3), also known as connexin 31 (Cx31) — is a protein that in humans is encoded by the GJB3 gene.

Delta-sarcoglycan is a protein that in humans is encoded by the SGCD gene.

Forkhead box protein E1 is a protein that in humans is encoded by the FOXE1 gene.

Homeobox protein MOX-1 is a protein that in humans is encoded by the MEOX1 gene.
Yemenite deaf-blind hypopigmentation syndrome is a condition caused by a mutation on the SRY-related HMG-box gene 10.

Gooseberry (gsb) is a segment polarity gene located on chromosome 2 of the Drosophila genome. Gooseberry is known for its interactions with key embryonic signaling pathways Wingless and Hedgehog. The gene also has clinal significance, being linked to diseases such as Waardenburg Syndrome and rhabdomyosarcoma.

Waardenburg syndrome type 2D, a subtype of the Waardenburg syndrome, is a rare congenital disorder caused by a mutation in the SLUG (SNAI2) gene. It is characterized by the lack of pigmentation in the skin, hair, and eyes as well as the abnormalities in the outer wall of the cochlea. This subtype lacks the wide distance between the eyes, known as dystopia canthorum, that is observed in most patients with Waardenburg syndrome. Those affected, exhibit varying degrees of deafness or complete hearing loss along with heterochromia and reports of early graying. This disease is observed in the neonatal stages of early life.

Waardenburg Syndrome Type 1 is a congenital disorder that caused by a mutation in the PAX3 gene that results in abnormal development in the neural crest during early development. Type 1 results in early graying and white forelock and a notable distance between the eyes, noted as dystopia canthorum. Common symptoms of the disease also includes non-progressive hearing loss in majority of patients with Type 1. Patients can display complete or partial heterochromia and hypoplastic blue irides and congenital leukemia.
Waardenburg Syndrome Type 4A is an extremely rare congenital disorder caused by a mutation in an endothelin receptor gene. It results in common Waardenburg syndrome symptoms such as abnormal hair and skin pigmentation and heterochromia, but also present with symptoms of Hirschsprung’s disease. Symptoms include abdominal pain and bowel obstruction. Waardenburg Syndrome Type 4A is the rarest among the types, appearing only once in about every 1,000,000 individuals. There have only been a total of 50 cases reported in total as of 2016.
This article incorporates text from the United States National Library of Medicine, which is in the public domain.