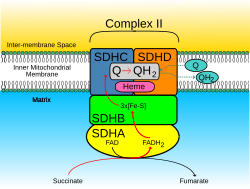
Clinical significance
Mutations in the SDHD gene can cause familial paraganglioma. [5] Germline mutations in SDHD were first linked to hereditary paraganglioma in 2000. [12] Since then, it has been shown that mutations in SDHB and to a lesser degree SDHC can cause paranglioma as well as familial pheochromocytoma. Notably, the tumor spectrum is different for the different mutations. SDHB mutations often lead to metastatic disease that is extra-adrenal, while SDHD mutation related tumors are more typically benign, originating in the head and neck. [13]
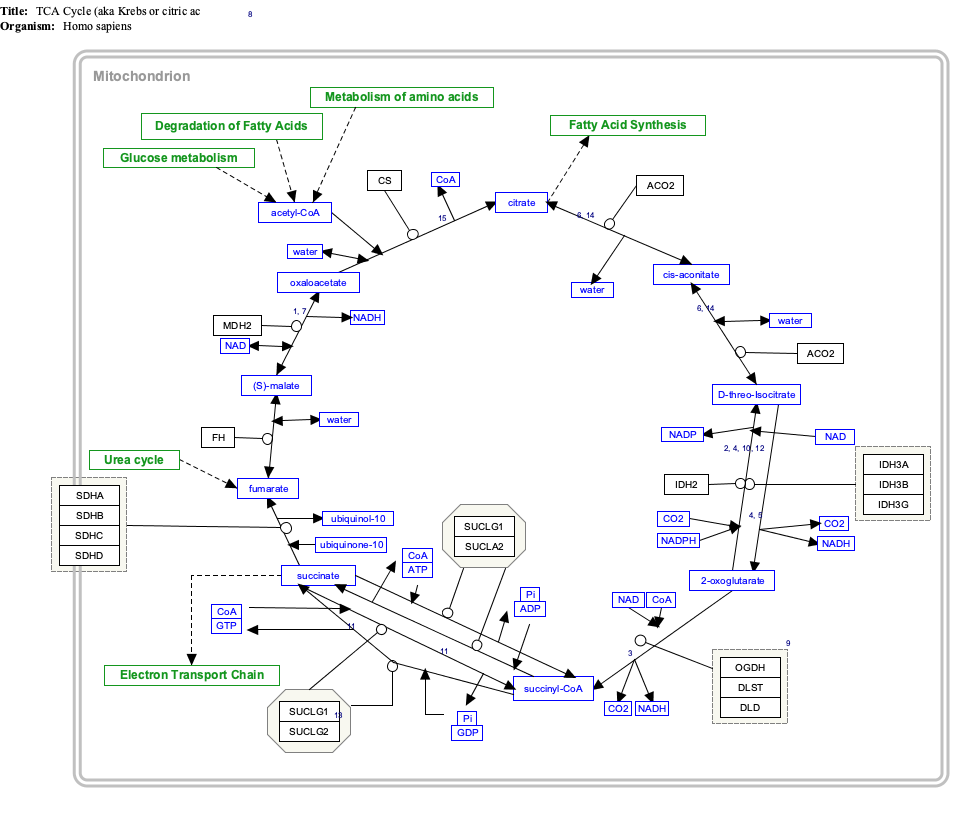
The exact mechanism for tumorigenesis is not determined, but it is suspected that malfunction of the SDH complex can cause a hypoxic response in the cell that leads to tumor formation. Mutations in the SDHB, SDHC, SDHD, and SDHAF2 genes lead to the loss or reduction of SDH enzyme activity. Because the mutated SDH enzyme cannot convert succinate to fumarate, succinate accumulates in the cell. As a result, the hypoxia pathways are triggered in normal oxygen conditions, which lead to abnormal cell growth and tumor formation. [13] People living at higher altitudes (for example, the Andes mountains) are known to have an increased rate of benign paraganglioma, with the rate of disease increasing with the altitude of the population.
At least five variants in the SDHD gene have been identified in people with Cowden syndrome or a similar disorder called Cowden-like syndrome. These conditions are characterized by multiple tumor-like growths called hamartomas and an increased risk of developing certain cancers. When Cowden syndrome and Cowden-like syndrome are caused by SDHD gene mutations, the conditions are associated with a particularly high risk of developing breast and thyroid cancers. The SDHD gene variants associated with Cowden syndrome and Cowden-like syndrome change single amino acids in the SDHD protein, which likely alters the function of the SDH enzyme. Studies suggest that the defective enzyme could allow cells to grow and divide unchecked, leading to the formation of hamartomas and cancerous tumors. However, researchers are uncertain whether the identified SDHD gene variants are directly associated with Cowden syndrome and Cowden-like syndrome. Some of the variants described above have rarely been found in people without the features of these conditions. [14]
Mutations in the SDHD gene have been found in a small number of people with Carney–Stratakis syndrome, a hereditary form of a cancer of the gastrointestinal tract called gastrointestinal stromal tumor (GIST). Those with Carney-Stratakis syndrome present with a noncancerous tumor associated with the nervous system called a paraganglioma or pheochromocytoma (a type of paraganglioma). An inherited SDHD gene mutation predisposes an individual to cancer formation. An additional mutation that deletes the normal copy of the gene is needed to cause Carney-Stratakis syndrome. This second mutation, called a somatic mutation, is acquired during a person's lifetime and is present only in tumor cells. [14]
Mitochondrial complex II deficiency (MT-C2D), a disorder of the mitochondrial respiratory chain with heterogeneous clinical manifestations, has also been associated with mutations in the SDHD gene. Clinical features include psychomotor regression in infants, poor growth with lack of speech development, severe spastic quadriplegia, dystonia, progressive leukoencephalopathy, muscle weakness, exercise intolerance, cardiomyopathy. Some patients manifest Leigh syndrome or Kearns–Sayre syndrome. [15] [16] [17]
This page is based on this
Wikipedia article Text is available under the
CC BY-SA 4.0 license; additional terms may apply.
Images, videos and audio are available under their respective licenses.