Dihydrolipoamide dehydrogenase (DLD), also known as dihydrolipoyl dehydrogenase, mitochondrial, is an enzyme that in humans is encoded by the DLDgene.[5][6][7][8] DLD is a flavoprotein enzyme that oxidizes dihydrolipoamide to lipoamide.
Dihydrolipoamide dehydrogenase (DLD) is a mitochondrial enzyme that plays a vital role in energy metabolism in eukaryotes. This enzyme is required for the complete reaction of at least five different multi-enzyme complexes.[9] Additionally, DLD is a flavoenzyme oxidoreductase that contains a reactive disulfide bridge and a FAD cofactor that are directly involved in catalysis. The enzyme associates into tightly bound homodimers required for its enzymatic activity.[10]
The protein encoded by the DLD gene comes together with another protein to form a dimer in the central metabolic pathway. Several amino acids within the catalytic pocket have been identified as important to DLD function, including R281 and N473.[11][12] Although the overall fold of the human enzyme is similar to that of yeast, the human structure is different in that it has two loops that extend from the general protein structure and into the FAD binding sites when bound the NAD+ molecule, required for catalysis, is not close to the FAD moiety. However, when NADH is bound instead, it is stacked directly op top of the FAD central structure. The current hE3 structures show directly that the disease-causing mutations occur at three locations in the human enzyme: the dimer interface, the active site, and the FAD and NAD(+)-binding sites.[13]
Function
The DLD homodimer functions as the E3 component of the pyruvate, α-ketoglutarate, α-adipate and branched-chain amino acid-dehydrogenase complexes and the glycine cleavage system, all in the mitochondrial matrix. In these complexes, DLD converts dihydrolipoic acid and NAD+ into lipoic acid and NADH.[14] DLD also has diaphorase activity, being able to catalyze the oxidation of NADH to NAD+ by using different electron acceptors such as O2, labile ferric iron, nitric oxide, and ubiquinone.[9] DLD is thought to have a pro-oxidant role by reducing oxygen to a superoxide or ferric to ferrous iron, which then catalyzes production of hydroxyl radicals.[15][16] Diaphorase activity of DLD may have an antioxidant role through its ability to scavenge nitric oxide and to reduce ubiquinone to ubiquinol.[17][18][19] The dihyrolipamide dehydrogenase gene is known to have multiple splice variants.
Moonlighting function
Certain DLD mutations can simultaneously induce the loss of a primary metabolic activity and the gain of a moonlighting proteolytic activity. The moonlighting proteolytic activity of DLD is revealed by conditions that destabilize the DLD homodimer and decrease its DLD activity.[9] Acidification of the mitochondrial matrix, as a result of ischemia-reperfusion injury, can disrupt the quaternary structure of DLD leading to decreased dehydrogenase activity and increased diaphorase activity.[20] The moonlighting proteolytic activity of DLD could also arise under pathological conditions. Proteolytic activity can further complicate the reduction in energy metabolism and an increase in oxidative damage as a result of decreased DLD activity and an increase in diaphorase activity respectively.[19] With its proteolytic function, DLD removes a functionally vital domain from the N-terminus of frataxin, a mitochondrial protein involved in iron metabolism and antioxidant protection.[21][22]
Clinical significance
In humans, mutations in DLD are linked to a severe disorder of infancy with failure to thrive, hypotonia, and metabolic acidosis. [23] DLD deficiency manifests itself in a great degree of variability, which has been attributed to varying effects of different DLD mutations on the stability of the protein and its ability to dimerize or interact with other components of the three α-ketoacid dehydrogenase complexes.[23] With its proteolytic function, DLD causes a deficiency in frataxin, which leads to the neurodegenerative and cardiac disease, Friedreich's ataxia.[24]
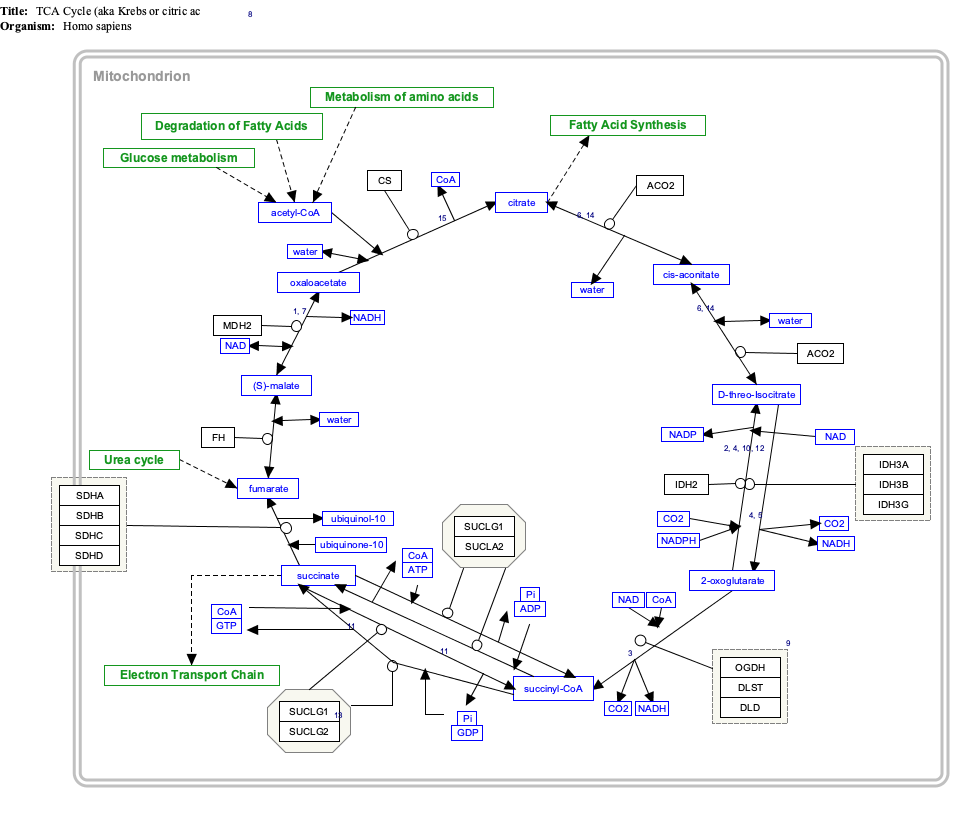
Interactive pathway map
Click on genes, proteins and metabolites below to link to respective articles.[§ 1]
↑ Wang YC, Wang ST, Li C, Chen LY, Liu WH, Chen PR, etal. (January 2008). "The role of amino acids T148 and R281 in human dihydrolipoamide dehydrogenase". Journal of Biomedical Science. 15 (1): 37–46. doi:10.1007/s11373-007-9208-9. PMID17960497.
↑ Brautigam CA, Chuang JL, Tomchick DR, Machius M, Chuang DT (July 2005). "Crystal structure of human dihydrolipoamide dehydrogenase: NAD+/NADH binding and the structural basis of disease-causing mutations". Journal of Molecular Biology. 350 (3): 543–52. doi:10.1016/j.jmb.2005.05.014. PMID15946682.
↑ Carothers DJ, Pons G, Patel MS (February 1989). "Dihydrolipoamide dehydrogenase: functional similarities and divergent evolution of the pyridine nucleotide-disulfide oxidoreductases". Archives of Biochemistry and Biophysics. 268 (2): 409–25. doi:10.1016/0003-9861(89)90309-3. PMID2643922.
↑ Yoneyama K, Shibata R, Igarashi A, Kojima S, Kodani Y, Nagata K, etal. (September 2014). "Proteomic identification of dihydrolipoamide dehydrogenase as a target of autoantibodies in patients with endometrial cancer". Anticancer Research. 34 (9): 5021–7. PMID25202086.
↑ O'Neill HA, Gakh O, Park S, Cui J, Mooney SM, Sampson M, etal. (January 2005). "Assembly of human frataxin is a mechanism for detoxifying redox-active iron". Biochemistry. 44 (2): 537–45. doi:10.1021/bi048459j. PMID15641778.
1 2 Quinonez SC, Thoene JG (9 July 2020). "Dihydrolipoamide Dehydrogenase Deficiency". In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Mirzaa G, Amemiya A (eds.). GeneReviews. University of Washington, Seattle. PMID25032271.
Brautigam CA, Chuang JL, Tomchick DR, Machius M, Chuang DT (July 2005). "Crystal structure of human dihydrolipoamide dehydrogenase: NAD+/NADH binding and the structural basis of disease-causing mutations". Journal of Molecular Biology. 350 (3): 543–52. doi:10.1016/j.jmb.2005.05.014. PMID15946682.
Asano K, Matsushita T, Umeno J, Hosono N, Takahashi A, Kawaguchi T, etal. (December 2009). "A genome-wide association study identifies three new susceptibility loci for ulcerative colitis in the Japanese population". Nature Genetics. 41 (12): 1325–9. doi:10.1038/ng.482. PMID19915573. S2CID20507558.
Odièvre MH, Chretien D, Munnich A, Robinson BH, Dumoulin R, Masmoudi S, etal. (March 2005). "A novel mutation in the dihydrolipoamide dehydrogenase E3 subunit gene (DLD) resulting in an atypical form of alpha-ketoglutarate dehydrogenase deficiency". Human Mutation. 25 (3): 323–4. doi:10.1002/humu.9319. PMID15712224. S2CID19929944.
Sugden MC, Holness MJ (May 2003). "Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by PDKs". American Journal of Physiology. Endocrinology and Metabolism. 284 (5): E855-62. doi:10.1152/ajpendo.00526.2002. PMID12676647.
Wang YC, Wang ST, Li C, Chen LY, Liu WH, Chen PR, etal. (January 2008). "The role of amino acids T148 and R281 in human dihydrolipoamide dehydrogenase". Journal of Biomedical Science. 15 (1): 37–46. doi:10.1007/s11373-007-9208-9. PMID17960497.
Brown AM, Gordon D, Lee H, Caudy M, Hardy J, Haroutunian V, Blass JP (November 2004). "Association of the dihydrolipoamide dehydrogenase gene with Alzheimer's disease in an Ashkenazi Jewish population". American Journal of Medical Genetics. Part B, Neuropsychiatric Genetics. 131B (1): 60–6. doi:10.1002/ajmg.b.30008. PMID15389771. S2CID26098296.
Wang YC, Wang ST, Li C, Liu WH, Chen PR, Chen LY, Liu TC (March 2007). "The role of N286 and D320 in the reaction mechanism of human dihydrolipoamide dehydrogenase (E3) center domain". Journal of Biomedical Science. 14 (2): 203–10. doi:10.1007/s11373-006-9136-0. PMID17171578.
Foster LJ, Rudich A, Talior I, Patel N, Huang X, Furtado LM, etal. (January 2006). "Insulin-dependent interactions of proteins with GLUT4 revealed through stable isotope labeling by amino acids in cell culture (SILAC)". Journal of Proteome Research. 5 (1): 64–75. doi:10.1021/pr0502626. PMID16396496.
1zy8: The crystal structure of dihydrolipoamide dehydrogenase and dihydrolipoamide dehydrogenase-binding protein (didomain) subcomplex of human pyruvate dehydrogenase complex.
2f5z: Crystal Structure of Human Dihydrolipoamide Dehydrogenase (E3) Complexed to the E3-Binding Domain of Human E3-Binding Protein
1zmc: Crystal Structure of Human dihydrolipoamide dehydrogenase complexed to NAD+
1zmd: Crystal Structure of Human dihydrolipoamide dehydrogenase complexed to NADH
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.