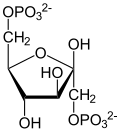
Phosphopyruvate hydratase belongs to the family of lyases, specifically the hydro-lyases, which cleave carbon-oxygen bonds. The systematic name of this enzyme is 2-phospho-D-glycerate hydro-lyase (phosphoenolpyruvate-forming).
The reaction is reversible, depending on environmental concentrations of substrates.[3] The optimum pH for the human enzyme is 6.5.[4] Enolase is present in all tissues and organisms capable of glycolysis or fermentation. The enzyme was discovered by Lohmann and Meyerhof in 1934,[5] and has since been isolated from a variety of sources including human muscle and erythrocytes.[4] In humans, deficiency of ENO1 is linked to hereditary haemolytic anemia, while ENO3 deficiency is linked to glycogen storage diseasetype XIII.
Isozymes
In humans there are three subunits of enolase, α, β, and γ, each encoded by a separate gene that can combine to form five different isoenzymes: αα, αβ, αγ, ββ, and γγ.[3][6] Three of these isoenzymes (all homodimers) are more commonly found in adult human cells than the others:
αα or non-neuronal enolase (NNE). Also known as enolase 1. Found in a variety of tissues, including liver, brain, kidney, spleen, adipose. It is present at some level in all normal human cells.
ββ or muscle-specific enolase (MSE). Also known as enolase 3. This enzyme is largely restricted to muscle tissue, where it is present at very high levels.
γγ or neuron-specific enolase (NSE). Also known as enolase 2. Expressed at very high levels in neurons and neural tissues, where it can account for as much as 3% of total soluble protein. It is expressed at much lower levels in most mammalian cells.
Enolase is a member of the large enolase superfamily. It has a molecular weight of 82,000–100,000 daltons depending on the isoform.[3][4] In human alpha enolase, the two subunits are antiparallel in orientation so that Glu20 of one subunit forms an ionic bond with Arg414 of the other subunit.[3] Each subunit has two distinct domains. The smaller N-terminal domain consists of three α-helices and four β-sheets.[3][6] The larger C-terminal domain starts with two β-sheets followed by two α-helices and ends with a barrel composed of alternating β-sheets and α-helices arranged so that the β-beta sheets are surrounded by the α-helices.[3][6] The enzyme's compact, globular structure results from significant hydrophobic interactions between these two domains.
Enolase is a highly conserved enzyme with five active-site residues being especially important for activity. When compared to wild-type enolase, a mutant enolase that differs at either the Glu168, Glu211, Lys345, or Lys396 residue has an activity level that is cut by a factor of 105.[3] Also, changes affecting His159 leave the mutant with only 0.01% of its catalytic activity.[3] An integral part of enolase are two Mg2+ cofactors in the active site, which serve to stabilize negative charges in the substrate.[3][6]
Recently, moonlighting functions of several enolases, such as interaction with plasminogen, have brought interest to the enzymes' catalytic loops and their structural diversity.[7][8]
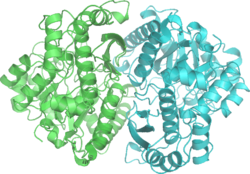
3-D depiction of enolase dimer in antiparallel orientation. One dimer's N-terminal Glu20 forms an ionic bond with the other's C-terminal Arg414 to stabilize the enzyme's quaternary structure.
Active site of enolase in the middle of the C-terminal domain's barrel. Depicted are two Mg2+ cofactors and five highly conserved residues imperative for proper catalytic function: His159, Glu168, Glu211, Lys345, Lys396.
Mechanism
Mechanism for conversion of 2PG to PEP.
Using isotopic probes, the overall mechanism for converting 2-PG to PEP is proposed to be an E1cB elimination reaction involving a carbanion intermediate.[9] The following detailed mechanism is based on studies of crystal structure and kinetics.[3][10][11][12][13][14][15] When the substrate, 2-phosphoglycerate, binds to α-enolase, its carboxyl group coordinates with two magnesium ion cofactors in the active site. This stabilizes the negative charge on the deprotonated oxygen while increasing the acidity of the alpha hydrogen. Enolase's Lys345 deprotonates the alpha hydrogen, and the resulting negative charge is stabilized by resonance to the carboxylate oxygen and by the magnesium ion cofactors. Following the creation of the carbanion intermediate, the hydroxide on C3 is eliminated as water with the help of Glu211, and PEP is formed.
Additionally, conformational changes occur within the enzyme that aid catalysis. In human α-enolase, the substrate is rotated into position upon binding to the enzyme due to interactions with the two catalytic magnesium ions, Gln167, and Lys396. Movements of loops Ser36 to His43, Ser158 to Gly162, and Asp255 to Asn256 allow Ser39 to coordinate with Mg2+ and close off the active site. In addition to coordination with the catalytic magnesium ions, the pKa of the substrate's alpha hydrogen is also lowered due to protonation of the phosphoryl group by His159 and its proximity to Arg374. Arg374 also causes Lys345 in the active site to become deprotonated, which primes Lys345 for its role in the mechanism.
Diagnostic uses
In recent medical experiments, enolase concentrations have been sampled in an attempt to diagnose certain conditions and their severity. For example, higher concentrations of enolase in cerebrospinal fluid more strongly correlated to low-grade astrocytoma than did other enzymes tested (aldolase, pyruvate kinase, creatine kinase, and lactate dehydrogenase).[16] The same study showed that the fastest rate of tumor growth occurred in patients with the highest levels of CSF enolase. Increased levels of enolase have also been identified in patients who have suffered a recent myocardial infarction or cerebrovascular accident. It has been inferred that levels of CSF neuron-specific enolase, serum NSE, and creatine kinase (type BB) are indicative in the prognostic assessment of cardiac arrest victims.[17] Other studies have focused on the prognostic value of NSE values in cerebrovascular accident victims.[18]
Small-molecule inhibitors of enolase have been synthesized as chemical probes (substrate-analogues) of the catalytic mechanism of the enzyme and more recently, have been investigated as potential treatments for cancer and infectious diseases.[21][22] Most inhibitors have metal chelating properties and bind to enzyme by interactions with the structural Magnesium Atom Mg(A).[23][24] The most potent of these is phosphonoacetohydroxamate,[24] which in its unprotonated form has pM affinity for the enzyme. It has structural similarity to the presumed catalytic intermediate, between PEP and 2-PG. Attempts have been made to use this inhibitor as an anti-trypanosome drug,[25] and more recently, as an anti-cancer agent, specifically, in glioblastoma that are enolase-deficient due to homozygous deletion of the ENO1 gene as part of the 1p36 tumor suppressor locus (synthetic lethality).[26] A natural product phosphonate antibiotic, SF2312 (CAS 107729-45-3), which is active against gram positive and negative bacteria especially under anaerobic conditions,[27] is a high potency inhibitor of Enolase 4zcw that binds in manner similar to phoshphonoacetohydroxamate 4za0.[28] SF2312 inhibits Enolase activity in both eukaryotic and prokaryotic origin,[29] reflecting the strong evolutionary conservation of Enolase and the ancient origin of the glycolysis pathway. SF2312 is a chiral molecule with only the 3S-enantiomer showing Enolase inhibitory activity and biological activity against bacteria.[30] More recently, a derivative of SF2312, termed HEX, and a prodrug thereof, POMHEX, were shown to exert anti-neoplastic activity against ENO1-deleted glioma in a pre-clinical intracranial orthotopic mouse model.[31] An allosteric binder, ENOblock[22] was initially described as an inhibitor of Enolase, but subsequently shown not to actually inhibit the enzyme, but rather, interfere with the Enolase in vitro enzymatic assay.[32] ENOblock was found to alter the cellular localization of enolase, influencing its secondary, non-glycolytic functions, such as transcription regulation.[33] Subsequent analysis using a commercial assay also indicated that ENOblock can inhibit enolase activity in biological contexts, such as cells and animal tissues.[33] Methylglyoxal has also been described as an inhibitor of human enolase.[34]
Active site transition state analogue Enolase inhibitors have been explored pre-clinically for the treatment of various microbial pathogens, as well as in precision oncology for tumors with 1p36 homozygous deletions, that lack ENO1.[31][35][36][37][38][39][40]
Fluoride is a known competitor of enolase's substrate 2-PG. Fluoride can form a complex with magnesium and phosphate, which binds in the active site instead of 2-PG.[4] One study found that fluoride could inhibit bacterial enolase in vitro.[41] The Enolase inhibitory activity of Fluoride anion may contribute to the anti-cavity effect of fluoride toothpaste, by limiting lactic acid (a product of glycolysis, which requires Enolase) production.[medical citation needed]
References
↑PDB: 2ONE; Zhang E, Brewer JM, Minor W, Carreira LA, Lebioda L (October 1997). "Mechanism of enolase: the crystal structure of asymmetric dimer enolase-2-phospho-D-glycerate/enolase-phosphoenolpyruvate at 2.0 A resolution". Biochemistry. 36 (41): 12526–12534. doi:10.1021/bi9712450. PMID9376357.
↑PDB: 2XSX; Vollmar M, Krysztofinska E, Chaikuad A, Krojer T, Cocking R, Vondelft F, Bountra C, Arrowsmith CH, Weigelt J, Edwards A, Yue WW, Oppermann U (2010). "Crystal structure of human beta enolase ENOB". Protein Data Bank. doi:10.2210/pdb2xsx/pdb.
↑Lohman, K; Meyerhof, O (1934). "Über die enzymatische umwandlung von phosphoglyzerinsäure in brenztraubensäure und phosphorsäure" [Enzymatic transformation of phosphoglyceric acid into pyruvic and phosphoric acid]. Biochemische Zeitschrift (in German). 273: 60–72.
↑Poyner RR, Laughlin LT, Sowa GA, Reed GH (February 1996). "Toward identification of acid/base catalysts in the active site of enolase: comparison of the properties of K345A, E168Q, and E211Q variants". Biochemistry. 35 (5): 1692–1699. doi:10.1021/bi952186y. PMID8634301.
↑Reed GH, Poyner RR, Larsen TM, Wedekind JE, Rayment I (December 1996). "Structural and mechanistic studies of enolase". Current Opinion in Structural Biology. 6 (6): 736–743. doi:10.1016/S0959-440X(96)80002-9. PMID8994873.
↑Wedekind JE, Reed GH, Rayment I (April 1995). "Octahedral coordination at the high-affinity metal site in enolase: crystallographic analysis of the MgII--enzyme complex from yeast at 1.9 A resolution". Biochemistry. 34 (13): 4325–4330. doi:10.1021/bi00013a022. PMID7703246.
↑Wedekind JE, Poyner RR, Reed GH, Rayment I (August 1994). "Chelation of serine 39 to Mg2+ latches a gate at the active site of enolase: structure of the bis(Mg2+) complex of yeast enolase and the intermediate analog phosphonoacetohydroxamate at 2.1-A resolution". Biochemistry. 33 (31): 9333–9342. doi:10.1021/bi00197a038. PMID8049235.
↑Larsen TM, Wedekind JE, Rayment I, Reed GH (April 1996). "A carboxylate oxygen of the substrate bridges the magnesium ions at the active site of enolase: structure of the yeast enzyme complexed with the equilibrium mixture of 2-phosphoglycerate and phosphoenolpyruvate at 1.8 A resolution". Biochemistry. 35 (14): 4349–4358. doi:10.1021/bi952859c. PMID8605183.
↑Lundberg K, Kinloch A, Fisher BA, Wegner N, Wait R, Charles P, etal. (October 2008). "Antibodies to citrullinated alpha-enolase peptide 1 are specific for rheumatoid arthritis and cross-react with bacterial enolase". Arthritis and Rheumatism. 58 (10): 3009–3019. doi:10.1002/art.23936. PMID18821669.
↑Fujii A, Yoneda M, Ito T, Yamamura O, Satomi S, Higa H, etal. (May 2005). "Autoantibodies against the amino terminal of alpha-enolase are a useful diagnostic marker of Hashimoto's encephalopathy". Journal of Neuroimmunology. 162 (1–2): 130–136. doi:10.1016/j.jneuroim.2005.02.004. PMID15833368. S2CID43249019.
↑Anderson VE, Weiss PM, Cleland WW (June 1984). "Reaction intermediate analogues for enolase". Biochemistry. 23 (12): 2779–2786. doi:10.1021/bi00307a038. PMID6380574.
12Jung DW, Kim WH, Park SH, Lee J, Kim J, Su D, etal. (2 April 2013). "A unique small molecule inhibitor of enolase clarifies its role in fundamental biological processes". ACS Chemical Biology. 8 (6): 1271–1282. doi:10.1021/cb300687k. PMID23547795.
↑Poyner RR, Reed GH (August 1992). "Structure of the bis divalent cation complex with phosphonoacetohydroxamate at the active site of enolase". Biochemistry. 31 (31): 7166–7173. doi:10.1021/bi00146a020. PMID1322695.
12Zhang E, Hatada M, Brewer JM, Lebioda L (May 1994). "Catalytic metal ion binding in enolase: the crystal structure of an enolase-Mn2+-phosphonoacetohydroxamate complex at 2.4-A resolution". Biochemistry. 33 (20): 6295–6300. doi:10.1021/bi00186a032. PMID8193144.
↑Watanabe H, Yoshida J, Tanaka E, Ito M, Miyadoh S, Shomura T (1986). "Studies on a new phosphonic acid antibiotic, SF-2312". Sci Rep Meiji Seika Kaisha. 25: 12–17.
↑Pietkiewicz J, Gamian A, Staniszewska M, Danielewicz R (April 2009). "Inhibition of human muscle-specific enolase by methylglyoxal and irreversible formation of advanced glycation end products". Journal of Enzyme Inhibition and Medicinal Chemistry. 24 (2): 356–364. doi:10.1080/14756360802187679. PMID18830874. S2CID85416928.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.

 3-D depiction of enolase dimer in antiparallel orientation. One dimer's N-terminal Glu20 forms an ionic bond with the other's C-terminal Arg414 to stabilize the enzyme's quaternary structure.
3-D depiction of enolase dimer in antiparallel orientation. One dimer's N-terminal Glu20 forms an ionic bond with the other's C-terminal Arg414 to stabilize the enzyme's quaternary structure. Active site of enolase in the middle of the C-terminal domain's barrel. Depicted are two Mg2+ cofactors and five highly conserved residues imperative for proper catalytic function: His159, Glu168, Glu211, Lys345, Lys396.
Active site of enolase in the middle of the C-terminal domain's barrel. Depicted are two Mg2+ cofactors and five highly conserved residues imperative for proper catalytic function: His159, Glu168, Glu211, Lys345, Lys396.