
Phenylalanine is an essential α-amino acid with the formula C
9H
11NO
2. It can be viewed as a benzyl group substituted for the methyl group of alanine, or a phenyl group in place of a terminal hydrogen of alanine. This essential amino acid is classified as neutral, and nonpolar because of the inert and hydrophobic nature of the benzyl side chain. The L-isomer is used to biochemically form proteins coded for by DNA. Phenylalanine is a precursor for tyrosine, the monoamine neurotransmitters dopamine, norepinephrine (noradrenaline), and epinephrine (adrenaline), and the biological pigment melanin. It is encoded by the messenger RNA codons UUU and UUC.
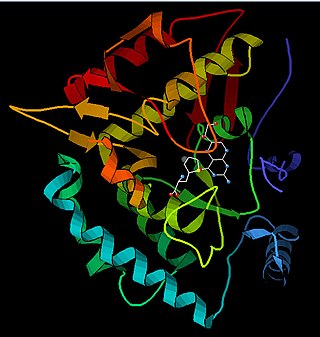
Phenylalanine hydroxylase (PAH) (EC 1.14.16.1) is an enzyme that catalyzes the hydroxylation of the aromatic side-chain of phenylalanine to generate tyrosine. PAH is one of three members of the biopterin-dependent aromatic amino acid hydroxylases, a class of monooxygenase that uses tetrahydrobiopterin (BH4, a pteridine cofactor) and a non-heme iron for catalysis. During the reaction, molecular oxygen is heterolytically cleaved with sequential incorporation of one oxygen atom into BH4 and phenylalanine substrate. In humans, mutations in its encoding gene, PAH, can lead to the metabolic disorder phenylketonuria.

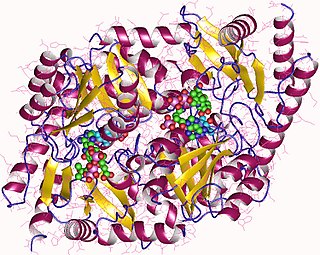
Pyruvate kinase is the enzyme involved in the last step of glycolysis. It catalyzes the transfer of a phosphate group from phosphoenolpyruvate (PEP) to adenosine diphosphate (ADP), yielding one molecule of pyruvate and one molecule of ATP. Pyruvate kinase was inappropriately named before it was recognized that it did not directly catalyze phosphorylation of pyruvate, which does not occur under physiological conditions. Pyruvate kinase is present in four distinct, tissue-specific isozymes in animals, each consisting of particular kinetic properties necessary to accommodate the variations in metabolic requirements of diverse tissues.
Aminolevulinic acid synthase (ALA synthase, ALAS, or delta-aminolevulinic acid synthase) is an enzyme (EC 2.3.1.37) that catalyzes the synthesis of δ-aminolevulinic acid (ALA) the first common precursor in the biosynthesis of all tetrapyrroles such as hemes, cobalamins and chlorophylls. The reaction is as follows:
Enoyl-CoA-(∆) isomerase (EC 5.3.3.8, also known as dodecenoyl-CoA- isomerase, 3,2-trans-enoyl-CoA isomerase, ∆3 ,∆2 -enoyl-CoA isomerase, or acetylene-allene isomerase, is an enzyme that catalyzes the conversion of cis- or trans-double bonds of coenzyme A bound fatty acids at gamma-carbon to trans double bonds at beta-carbon as below:

Pyridoxal phosphate (PLP, pyridoxal 5'-phosphate, P5P), the active form of vitamin B6, is a coenzyme in a variety of enzymatic reactions. The International Union of Biochemistry and Molecular Biology has catalogued more than 140 PLP-dependent activities, corresponding to ~4% of all classified activities. The versatility of PLP arises from its ability to covalently bind the substrate, and then to act as an electrophilic catalyst, thereby stabilizing different types of carbanionic reaction intermediates.
In biochemistry and metabolism, beta oxidation (also β-oxidation) is the catabolic process by which fatty acid molecules are broken down in the cytosol in prokaryotes and in the mitochondria in eukaryotes to generate acetyl-CoA. Acetyl-CoA enters the citric acid cycle, generating NADH and FADH2, which are electron carriers used in the electron transport chain. It is named as such because the beta carbon of the fatty acid chain undergoes oxidation and is converted to a carbonyl group to start the cycle all over again. Beta-oxidation is primarily facilitated by the mitochondrial trifunctional protein, an enzyme complex associated with the inner mitochondrial membrane, although very long chain fatty acids are oxidized in peroxisomes.
Tyrosine hydroxylase or tyrosine 3-monooxygenase is the enzyme responsible for catalyzing the conversion of the amino acid L-tyrosine to L-3,4-dihydroxyphenylalanine (L-DOPA). It does so using molecular oxygen (O2), as well as iron (Fe2+) and tetrahydrobiopterin as cofactors. L-DOPA is a precursor for dopamine, which, in turn, is a precursor for the important neurotransmitters norepinephrine (noradrenaline) and epinephrine (adrenaline). Tyrosine hydroxylase catalyzes the rate limiting step in this synthesis of catecholamines. In humans, tyrosine hydroxylase is encoded by the TH gene, and the enzyme is present in the central nervous system (CNS), peripheral sympathetic neurons and the adrenal medulla. Tyrosine hydroxylase, phenylalanine hydroxylase and tryptophan hydroxylase together make up the family of aromatic amino acid hydroxylases (AAAHs).

Trifunctional enzyme subunit alpha, mitochondrial also known as hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase/enoyl-CoA hydratase, alpha subunit is a protein that in humans is encoded by the HADHA gene. Mutations in HADHA have been associated with trifunctional protein deficiency or long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency.
Serine dehydratase or L-serine ammonia lyase (SDH) is in the β-family of pyridoxal phosphate-dependent (PLP) enzymes. SDH is found widely in nature, but its structure and properties vary among species. SDH is found in yeast, bacteria, and the cytoplasm of mammalian hepatocytes. SDH catalyzes the deamination of L-serine to yield pyruvate, with the release of ammonia.

Aminolevulinic acid dehydratase (porphobilinogen synthase, or ALA dehydratase, or aminolevulinate dehydratase) is an enzyme (EC 4.2.1.24) that in humans is encoded by the ALAD gene. Porphobilinogen synthase (or ALA dehydratase, or aminolevulinate dehydratase) synthesizes porphobilinogen through the asymmetric condensation of two molecules of aminolevulinic acid. All natural tetrapyrroles, including hemes, chlorophylls and vitamin B12, share porphobilinogen as a common precursor. Porphobilinogen synthase is the prototype morpheein.
The enzyme L-serine ammonia-lyase (EC 4.3.1.17) catalyzes the chemical reaction
The enzyme 4a-hydroxytetrahydrobiopterin dehydratase (EC 4.2.1.96) catalyzes the chemical reaction

Arogenate dehydratase (ADT) (EC 4.2.1.91) is an enzyme that catalyzes the chemical reaction
In enzymology, glutamate-prephenate aminotransferase is an enzyme that catalyzes the chemical reaction

D-bifunctional protein (DBP), also known as peroxisomal multifunctional enzyme type 2 (MFP-2), as well as 17β-hydroxysteroid dehydrogenase type IV is a protein that in humans is encoded by the HSD17B4 gene. It's an alcohol oxidoreductase, specifically 17β-Hydroxysteroid dehydrogenase. It is involved in fatty acid β-oxidation and steroid metabolism.

Pterin-4-alpha-carbinolamine dehydratase is an enzyme that in humans is encoded by the PCBD1 gene.

Lathosterol oxidase is a Δ7-sterol 5(6)-desaturase enzyme that in humans is encoded by the SC5D gene.

Hydroxyacyl-Coenzyme A dehydrogenase (HADH) is an enzyme which in humans is encoded by the HADH gene.

Biopterin-dependent aromatic amino acid hydroxylases (AAAH) are a family of aromatic amino acid hydroxylase enzymes which includes phenylalanine 4-hydroxylase, tyrosine 3-hydroxylase, and tryptophan 5-hydroxylase. These enzymes primarily hydroxylate the amino acids L-phenylalanine, L-tyrosine, and L-tryptophan, respectively.
