
Phenylketonuria (PKU) is an inborn error of metabolism that results in decreased metabolism of the amino acid phenylalanine. Untreated PKU can lead to intellectual disability, seizures, behavioral problems, and mental disorders. It may also result in a musty smell and lighter skin. A baby born to a mother who has poorly treated PKU may have heart problems, a small head, and low birth weight.
Phenylalanine is an essential α-amino acid with the formula C
9H
11NO
2. It can be viewed as a benzyl group substituted for the methyl group of alanine, or a phenyl group in place of a terminal hydrogen of alanine. This essential amino acid is classified as neutral, and nonpolar because of the inert and hydrophobic nature of the benzyl side chain. The L-isomer is used to biochemically form proteins coded for by DNA. Phenylalanine is a precursor for tyrosine, the monoamine neurotransmitters dopamine, norepinephrine (noradrenaline), and epinephrine (adrenaline), and the biological pigment melanin. It is encoded by the messenger RNA codons UUU and UUC.

The neonatal heel prick is a blood collection procedure done on newborns. It consists of making a pinprick puncture in one heel of the newborn to collect their blood. This technique is used frequently as the main way to collect blood from neonates. Other techniques include venous or arterial needle sticks, cord blood sampling, or umbilical line collection. This technique is often utilized for the Guthrie test, where it is used to soak the blood into pre-printed collection cards known as Guthrie cards.

A catecholamine is a monoamine neurotransmitter, an organic compound that has a catechol and a side-chain amine.
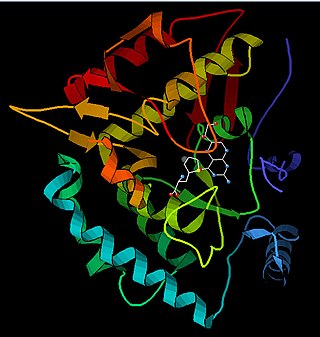
Phenylalanine hydroxylase (PAH) (EC 1.14.16.1) is an enzyme that catalyzes the hydroxylation of the aromatic side-chain of phenylalanine to generate tyrosine. PAH is one of three members of the biopterin-dependent aromatic amino acid hydroxylases, a class of monooxygenase that uses tetrahydrobiopterin (BH4, a pteridine cofactor) and a non-heme iron for catalysis. During the reaction, molecular oxygen is heterolytically cleaved with sequential incorporation of one oxygen atom into BH4 and phenylalanine substrate. In humans, mutations in its encoding gene, PAH, can lead to the metabolic disorder phenylketonuria.

Tetrahydrobiopterin (BH4, THB), also known as sapropterin (INN), is a cofactor of the three aromatic amino acid hydroxylase enzymes, used in the degradation of amino acid phenylalanine and in the biosynthesis of the neurotransmitters serotonin (5-hydroxytryptamine, 5-HT), melatonin, dopamine, norepinephrine (noradrenaline), epinephrine (adrenaline), and is a cofactor for the production of nitric oxide (NO) by the nitric oxide synthases. Chemically, its structure is that of a (dihydropteridine reductase) reduced pteridine derivative (quinonoid dihydrobiopterin).
Tetrahydrobiopterin deficiency (THBD, BH4D) is a rare metabolic disorder that increases the blood levels of phenylalanine. Phenylalanine is an amino acid obtained normally through the diet, but can be harmful if excess levels build up, causing intellectual disability and other serious health problems. In healthy individuals, it is metabolised (hydroxylated) into tyrosine, another amino acid, by phenylalanine hydroxylase. However, this enzyme requires tetrahydrobiopterin as a cofactor and thus its deficiency slows phenylalanine metabolism.

6-Pyruvoyltetrahydropterin synthase deficiency is an autosomal recessive disorder that causes malignant hyperphenylalaninemia due to tetrahydrobiopterin deficiency. It is a recessive disorder that is accompanied by hyperphenylalaninemia. Commonly reported symptoms are initial truncal hypotonia, subsequent appendicular hypertonia, bradykinesia, cogwheel rigidity, generalized dystonia, and marked diurnal fluctuation. Other reported clinical features include difficulty in swallowing, oculogyric crises, somnolence, irritability, hyperthermia, and seizures. Chorea, athetosis, hypersalivation, rash with eczema, and sudden death have also been reported. Patients with mild phenotypes may deteriorate if given folate antagonists such as methotrexate, which can interfere with a salvage pathway through which dihydrobiopterin is converted into tetrahydrobiopterin via dihydrofolate reductase. Treatment options include substitution with neurotransmitter precursors, monoamine oxidase inhibitors, and tetrahydrobiopterin. Response to treatment is variable and the long-term and functional outcome is unknown. To provide a basis for improving the understanding of the epidemiology, genotype–phenotype correlation and outcome of these diseases, their impact on the quality of life of patients, and for evaluating diagnostic and therapeutic strategies a patient registry was established by the noncommercial International Working Group on Neurotransmitter Related Disorders (iNTD).

GTP cyclohydrolase I (GTPCH) (EC 3.5.4.16) is a member of the GTP cyclohydrolase family of enzymes. GTPCH is part of the folate and biopterin biosynthesis pathways. It is responsible for the hydrolysis of guanosine triphosphate (GTP) to form 7,8-dihydroneopterin triphosphate (7,8-DHNP-3'-TP, 7,8-NH2-3'-TP).
Robert Guthrie, MD, Ph.D. was an American microbiologist, best known for developing the bacterial inhibition assay used to screen infants for phenylketonuria at birth, before the development of irreversible neurological damage. Guthrie also pioneered the collection of whole blood on specially designed filter paper, commonly known as "Guthrie cards" as a sample medium that could be easily collected, transported and tested. Although Guthrie is best known for developing the test for phenylketonuria, he worked tirelessly to raise awareness of the need to screen for treatable conditions and adapted his method to early screening tests for galactosemia and maple syrup urine disease.

Tryptophan hydroxylase (TPH) is an enzyme (EC 1.14.16.4) involved in the synthesis of the monoamine neurotransmitter serotonin. Tyrosine hydroxylase, phenylalanine hydroxylase, and tryptophan hydroxylase together constitute the family of biopterin-dependent aromatic amino acid hydroxylases. TPH catalyzes the following chemical reaction

The enzyme phenylalanine racemase is the enzyme that acts on amino acids and derivatives. It activates both the L & D stereo isomers of phenylalanine to form L-phenylalanyl adenylate and D-phenylalanyl adenylate, which are bound to the enzyme. These bound compounds are then transferred to the thiol group of the enzyme followed by conversion of its configuration, the D-isomer being the more favorable configuration of the two, with a 7 to 3 ratio between the two isomers. The racemisation reaction of phenylalanine is coupled with the highly favorable hydrolysis of adenosine triphosphate (ATP) to adenosine monophosphate (AMP) and pyrophosphate (PP), thermodynamically allowing it to proceed. This reaction is then drawn forward by further hydrolyzing PP to inorganic phosphate (Pi), via Le Chatelier's principle.

The enzyme phenylalanine ammonia lyase (EC 4.3.1.24) catalyzes the conversion of L-phenylalanine to ammonia and trans-cinnamic acid.:
Dopamine-responsive dystonia (DRD) also known as Segawa syndrome (SS), is a genetic movement disorder which usually manifests itself during early childhood at around ages 5–8 years.

Biopterin-dependent aromatic amino acid hydroxylases (AAAH) are a family of aromatic amino acid hydroxylase enzymes which includes phenylalanine 4-hydroxylase, tyrosine 3-hydroxylase, and tryptophan 5-hydroxylase. These enzymes primarily hydroxylate the amino acids L-phenylalanine, L-tyrosine, and L-tryptophan, respectively.

Pegvaliase, sold under the brand name Palynziq, is a medication used for the treatment of the genetic disease phenylketonuria. It is a phenylalanine (Phe)‑metabolizing enzyme. Chemically, it is a pegylated derivative of the enzyme phenylalanine ammonia-lyase that metabolizes phenylalanine to reduce its blood levels.
Harvey Louis Levy is an American biochemical geneticist, pediatrician, physician scientist and academic. He is Senior Physician in Medicine and Genetics at Boston Children’s Hospital and Professor of Pediatrics at Harvard Medical School.
Dihydropteridine reductase deficiency (DHPRD) is a genetic disorder affecting the tetrahydrobiopterin (BH4) synthesis pathway, inherited in the autosomal recessive pattern. It is one of the six known disorders causing tetrahydrobiopterin deficiency, and occurs in patients with mutations of the QDPR gene.
Autosomal recessive GTP cyclohydrolase I deficiency (AR-GTPCHD) is a disorder associated with the deficient operation of the enzyme GTP cyclohydrolase I. The condition leads to insufficient production of the cofactor tetrahydrobiopterin necessary for the proper synthesis of dopamine and serotonin and for maintenance of adequate levels of phenylalanine. As of 2020, autosomal recessive GTP cyclohydrolase I deficiency was one of the six known causes of tetrahydrobiopterin deficiency. It is also considered part of the spectrum of dopa-responsive dystonias.

Louis Isaac Woolf was a British biochemist who played a crucial role in the early detection and the treatment of phenylketonuria (PKU).
