The reaction is thought to proceed through the following steps:
formation of a Fe(II)-O-O-BH4 bridge.
heterolytic cleavage of the O-O bond to yield the ferryl oxo hydroxylating intermediate Fe(IV)=O
attack on Fe(IV)=O to hydroxylate phenylalanine substrate to tyrosine.[7]
PAH mechanism, part I
Formation and cleavage of the iron-peroxypterin bridge. Although evidence strongly supports Fe(IV)=O as the hydroxylating intermediate,[8] the mechanistic details underlying the formation of the Fe(II)-O-O-BH4 bridge prior to heterolytic cleavage remain controversial. Two pathways have been proposed based on models that differ in the proximity of the iron to the pterin cofactor and the number of water molecules assumed to be iron-coordinated during catalysis. According to one model, an iron dioxygen complex is initially formed and stabilized as a resonance hybrid of Fe2+O2 and Fe3+O2−. The activated O2 then attacks BH4, forming a transition state characterized by charge separation between the electron-deficient pterin ring and the electron-rich dioxygen species.[9] The Fe(II)-O-O-BH4 bridge is subsequently formed. On the other hand, formation of this bridge has been modeled assuming that BH4 is located in iron's first coordination shell and that the iron is not coordinated to any water molecules. This model predicts a different mechanism involving a pterin radical and superoxide as critical intermediates.[10] Once formed, the Fe(II)-O-O-BH4 bridge is broken through heterolytic cleavage of the O-O bond to Fe(IV)=O and 4a-hydroxytetrahydrobiopterin; thus, molecular oxygen is the source of both oxygen atoms used to hydroxylate the pterin ring and phenylalanine.
PAH mechanism, part II
Hydroxylation of phenylalanine by ferryl oxo intermediate. Because the mechanism involves a Fe(IV)=O (as opposed to a peroxypterin) hydroxylating intermediate, oxidation of the BH4 cofactor and hydroxylation of phenylalanine can be decoupled, resulting in unproductive consumption of BH4 and formation of H2O2.[7] When productive, though, the Fe(IV)=O intermediate is added to phenylalanine in an electrophilic aromatic substitution reaction that reduces iron from the ferryl to the ferrous state.[7] Although initially an arene oxide or radical intermediate was proposed, analyses of the related tryptophan and tyrosine hydroxylases have suggested that the reaction instead proceeds through a cationic intermediate that requires Fe(IV)=O to be coordinated to a water ligand rather than a hydroxo group.[7][11] This cationic intermediate subsequently undergoes a 1,2-hydride NIH shift, yielding a dienone intermediate that then tautomerizes to form the tyrosine product.[12] The pterin cofactor is regenerated by hydration of the carbinolamine product of PAH to quinonoid dihydrobiopterin (qBH2), which is then reduced to BH4.[13]
Mammalian PAH exists in an equilibrium consisting of tetramers of two distinct architectures, with one or more dimeric forms as part of the equilibrium. This behavior is consistent with a dissociative allosteric mechanism.[15]
Many studies suggest that mammalian PAH shows behavior comparable to porphobilinogen synthase (PBGS), wherein a variety of factors such as pH and ligand binding are reported to affect enzyme activity and protein stability.[15]
Structure
The PAH monomer (51.9 kDa) consists of three distinct domains: a regulatory N-terminal domain (residues 1–117) that contains a Phe-binding ACT subdomain, the catalytic domain (residues 118–427), and a C-terminal domain (residues 428–453) responsible for oligomerization of identical monomers. Extensive crystallographic analysis has been performed, especially on the pterin- and iron-coordinated catalytic domain to examine the active site. The structure of the N-terminal regulatory domain has also been determined, and together with the solved structure of the homologous tyrosine hydroxylase C-terminal tetramerization domain, a structural model of tetrameric PAH was proposed.[13] Using X-ray crystallography, the structure of full-length rat PAH was determined experimentally and showed the auto-inhibited, or resting-state form of the enzyme.[16] The resting-state form (RS-PAH) is architecturally distinct from the activated form (A-PAH).[17] A full-length structure of A-PAH is currently lacking, but the Phe stabilized ACT-ACT interface that is characteristic of A-PAH has been determined and a structural model of A-PAH based on SAXS analysis has been proposed.[18][19]
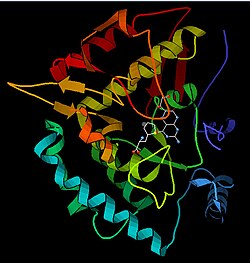
Model of the active site of PAH bound to BH4, ferrous, and a phenylalanine analogue. (from PDB 1KW0) Phenylalanine analogue, BH4, iron, Fe(II)-coordinated His and Glu residues
Catalytic domain
Solved crystal structures of the catalytic domain indicate that the active site consists of an open and spacious pocket lined primarily by hydrophobic residues, though three glutamic acid residues, two histidines, and a tyrosine are also present and iron-binding.[13] Contradictory evidence exists about the coordination state of the ferrous atom and its proximity to BH4 within the active site. According to crystallographic analysis, Fe(II) is coordinated by water, His285, His290, and Glu330 (a 2-his-1-carboxylate facial triad arrangement) with octahedral geometry.[20] Inclusion of a Phe analogue in the crystal structure changes both iron from a six- to a five-coordinated state involving a single water molecule and bidentate coordination to Glu330 and opening a site for oxygen to bind. BH4 is concomitantly shifted toward the iron atom, although the pterin cofactor remains in the second coordination sphere.[21] On the other hand, a competing model based on NMR and molecular modeling analyses suggests that all coordinated water molecules are forced out of the active site during the catalytic cycle while BH4 becomes directly coordinated to iron.[22] As discussed above, resolving this discrepancy will be important for determining the exact mechanism of PAH catalysis.
N-terminal regulatory domain
The regulatory nature of the N-terminal domain (residues 1–117) is conferred by its structural flexibility.[23] Hydrogen/deuterium exchanges analysis indicates that allosteric binding of Phe globally alters the conformation of PAH such that the active site is less occluded as the interface between the regulatory and catalytic domains is increasingly exposed to solvent.[23][24][25] This observation is consistent with kinetic studies, which show an initially low rate of tyrosine formation for full-length PAH. This lag time is not observed, however, for a truncated PAH lacking the N-terminal domain or if the full-length enzyme is pre-incubated with Phe. Deletion of the N-terminal domain also eliminates the lag time while increasing the affinity for Phe by nearly two-fold; no difference is observed in the Vmax or Km for the tetrahydrobiopterin cofactor.[26] Additional regulation is provided by Ser16; phosphorylation of this residue does not alter enzyme conformation but does reduce the concentration of Phe required for allosteric activation.[25] This N-terminal regulatory domain is not observed in bacterial PAHs but shows considerable structural homology to the regulatory domain of phosphogylcerate dehydrogenase, an enzyme in the serine biosynthetic pathway.[25]
Tetramerization domain
Prokaryotic PAH is monomeric, whereas eukaryotic PAH exists in an equilibrium between homotetrameric and homodimeric forms.[7][13] The dimerization interface is composed of symmetry-related loops that link identical monomers, while the overlapping C-terminal tetramerization domain mediates the association of conformationally distinct dimers that are characterized by a different relative orientation of the catalytic and tetramerization domains (Flatmark, Erlandsen). The resulting distortion of the tetramer symmetry is evident in the differential surface area of the dimerization interfaces and distinguishes PAH from the tetramerically symmetrical tyrosine hydroxylase.[13] A domain-swapping mechanism has been proposed to mediate formation of the tetramer from dimers, in which C-terminal alpha-helixes mutually alter their conformation around a flexible C-terminal five-residue hinge region to form a coiled-coil structure, shifting equilibrium toward the tetrameric form.[7][13][27] Although both the homodimeric and homotetrameric forms of PAH are catalytically active, the two exhibit differential kinetics and regulation. In addition to reduced catalytic efficiency, the dimer does not display positive cooperativity toward L-Phe (which at high concentrations activates the enzyme), suggesting that L-Phe allosterically regulates PAH by influencing dimer-dimer interaction.[27]
Biological function
PAH is a critical enzyme in phenylalanine metabolism and catalyzes the rate-limiting step in its complete catabolism to carbon dioxide and water.[13][28] Regulation of flux through phenylalanine-associated pathways is critical in mammalian metabolism, as evidenced by the toxicity of high plasma levels of this amino acid observed in phenylketonuria (see below). The principal source of phenylalanine is ingested proteins, but relatively little of this pool is used for protein synthesis.[28] Instead, the majority of ingested phenylalanine is catabolized through PAH to form tyrosine; addition of the hydroxyl group allows for the benzene ring to be broken in subsequent catabolic steps. Transamination to phenylpyruvate, whose metabolites are excreted in the urine, represents another pathway of phenylalanine turnover, but catabolism through PAH predominates.[28]
In humans, this enzyme is expressed both in the liver and the kidney, and there is some indication that it may be differentially regulated in these tissues.[29] PAH is unusual among the aromatic amino acid hydroxylases for its involvement in catabolism; tyrosine and tryptophan hydroxylases, on the other hand, are primarily expressed in the central nervous system and catalyze rate-limiting steps in neurotransmitter/hormone biosynthesis.[13]
Disease relevance
Deficiency in PAH activity due to mutations in PAH causes hyperphenylalaninemia (HPA), and when blood phenylalanine levels increase above 20 times the normal concentration, the metabolic disease phenylketonuria (PKU) results.[28] PKU is both genotypically and phenotypically heterogeneous: Over 300 distinct pathogenic variants have been identified, the majority of which correspond to missense mutations that map to the catalytic domain.[13][20] When a cohort of identified PAH mutants were expressed in recombinant systems, the enzymes displayed altered kinetic behavior and/or reduced stability, consistent with structural mapping of these mutations to both the catalytic and tetramerization domains of the enzyme.[13] BH44 has been administered as a pharmacological treatment and has been shown to reduce blood levels of phenylalanine for a segment of PKU patients whose genotypes lead to some residual PAH activity but have no defect in BH44 synthesis or regeneration. Follow-up studies suggest that in the case of certain PAH mutants, excess BH44 acts as a pharmacological chaperone to stabilize mutant enzymes with disrupted tetramer assembly and increased sensitivity to proteolytic cleavage and aggregation.[30] Mutations that have been identified in the PAH locus are documented at the Phenylalanine Hydroxylase Locus Knowledgbase (PAHdb, https://web.archive.org/web/20130718162051/http://www.pahdb.mcgill.ca/).
Since phenylketonuria can cause irreversible damage, it is imperative that deficiencies in the phenylalanine hydroxylase are determined early on in development. Originally, this was done using a bacterial inhibition assay known as the Guthrie Test. Now, PKU is part of newborn screening in many countries, and elevated phenylalanine levels are identified shortly after birth by measurement with tandem mass spectrometry. Placing the individual on a low phenylalanine, high tyrosine diet can help prevent any long-term damage to their development.
PAH Knock-out Model
The first attempt at creating a Pah-KO mouse model was reported in a research article published in 2021.[31] This knockout mouse was created to be homozygous through its development within the C57BL/6 J strain using CRISPR/Cas9.[32] Codon 7, GAG, in the Pah gene was altered to the stop codon TAG, depicting an intentional point mutation. Two to six-month-old, male, homozygous mice were studied by scientific methods such as behavioral and biochemical assays, MRI, and histopathology. The homozygous mice were routinely compared to control mice, age and sex-matched heterozygous Pah-KO mice that expressed sufficient PAH enzyme activity to supply phenylalanine and tyrosine levels similar to wild type mice.
The Pah-KO mouse model presented high blood phenylalanine and low tyrosine levels, hypocholesterolemia, high phenylalanine and low levels of neurotransmitters and tyrosine in the brain, hypomyelination, low brain and body weight, high grade of ocular pathology, hypopigmentation, and a progressive behavioral deficit compared to heterozygous mice.[31] After termination of the homozygous mice, no hepatic PAH proteins were detected by western blot analysis.[33] The increased amount of phenylalanine in whole brain homogenates of homozygous mice was similar to PKU patients. The hair coat color of both mice was dependent on the amount of melanin pigment in hair shafts; thus, homozygous mice were prone to be a lighter brown based on less melanin present. Mice behavior was tested with a nesting building assay.[31]
This model sets up the potential for an extensive investigation of PKU biology resembling PKU patients and for the evaluation of modern therapeutic strategies for PKU treatment.
↑ Olsson E, Martinez A, Teigen K, Jensen VR (March 2011). "Formation of the iron-oxo hydroxylating species in the catalytic cycle of aromatic amino acid hydroxylases". Chemistry: A European Journal. 17 (13): 3746–58. Bibcode:2011ChEuJ..17.3746O. doi:10.1002/chem.201002910. PMID21351297.
↑ Bassan A, Blomberg MR, Siegbahn PE (September 2003). "Mechanism of aromatic hydroxylation by an activated FeIV=O core in tetrahydrobiopterin-dependent hydroxylases". Chemistry: A European Journal. 9 (17): 4055–67. doi:10.1002/chem.200304768. PMID12953191.
1 2 Erlandsen H, Fusetti F, Martinez A, Hough E, Flatmark T, Stevens RC (December 1997). "Crystal structure of the catalytic domain of human phenylalanine hydroxylase reveals the structural basis for phenylketonuria". Nature Structural Biology. 4 (12): 995–1000. doi:10.1038/nsb1297-995. PMID9406548. S2CID6293946.
↑ Andersen OA, Flatmark T, Hough E (July 2002). "Crystal structure of the ternary complex of the catalytic domain of human phenylalanine hydroxylase with tetrahydrobiopterin and 3-(2-thienyl)-L-alanine, and its implications for the mechanism of catalysis and substrate activation". Journal of Molecular Biology. 320 (5): 1095–108. doi:10.1016/S0022-2836(02)00560-0. PMID12126628.
↑ Teigen K, Frøystein NA, Martínez A (December 1999). "The structural basis of the recognition of phenylalanine and pterin cofactors by phenylalanine hydroxylase: implications for the catalytic mechanism". Journal of Molecular Biology. 294 (3): 807–23. doi:10.1006/jmbi.1999.3288. PMID10610798.
1 2 3 Kobe B, Jennings IG, House CM, Michell BJ, Goodwill KE, Santarsiero BD, etal. (May 1999). "Structural basis of autoregulation of phenylalanine hydroxylase". Nature Structural Biology. 6 (5): 442–8. doi:10.1038/8247. PMID10331871. S2CID11709986.
↑ Daubner SC, Hillas PJ, Fitzpatrick PF (December 1997). "Expression and characterization of the catalytic domain of human phenylalanine hydroxylase". Archives of Biochemistry and Biophysics. 348 (2): 295–302. doi:10.1006/abbi.1997.0435. PMID9434741.
1 2 Bjørgo E, de Carvalho RM, Flatmark T (February 2001). "A comparison of kinetic and regulatory properties of the tetrameric and dimeric forms of wild-type and Thr427→Pro mutant human phenylalanine hydroxylase: contribution of the flexible hinge region Asp425-Gln429 to the tetramerization and cooperative substrate binding". European Journal of Biochemistry. 268 (4): 997–1005. doi:10.1046/j.1432-1327.2001.01958.x. PMID11179966.
↑ Lichter-Konecki U, Hipke CM, Konecki DS (August 1999). "Human phenylalanine hydroxylase gene expression in kidney and other nonhepatic tissues". Molecular Genetics and Metabolism. 67 (4): 308–16. doi:10.1006/mgme.1999.2880. PMID10444341.
↑ Muntau AC, Gersting SW (December 2010). "Phenylketonuria as a model for protein misfolding diseases and for the development of next generation orphan drugs for patients with inborn errors of metabolism". Journal of Inherited Metabolic Disease. 33 (6): 649–58. doi:10.1007/s10545-010-9185-4. PMID20824346. S2CID20843095.
Konecki DS, Lichter-Konecki U (August 1991). "The phenylketonuria locus: current knowledge about alleles and mutations of the phenylalanine hydroxylase gene in various populations". Human Genetics. 87 (4): 377–88. doi:10.1007/BF00197152. PMID1679029. S2CID25627287.
Cotton RG (1991). "Heterogeneity of phenylketonuria at the clinical, protein and DNA levels". Journal of Inherited Metabolic Disease. 13 (5): 739–50. doi:10.1007/BF01799577. PMID2246858. S2CID21931016.
Erlandsen H, Fusetti F, Martinez A, Hough E, Flatmark T, Stevens RC (December 1997). "Crystal structure of the catalytic domain of human phenylalanine hydroxylase reveals the structural basis for phenylketonuria". Nature Structural Biology. 4 (12): 995–1000. doi:10.1038/nsb1297-995. PMID9406548. S2CID6293946.
Waters PJ (April 2003). "How PAH gene mutations cause hyper-phenylalaninemia and why mechanism matters: insights from in vitro expression". Human Mutation. 21 (4): 357–69. doi:10.1002/humu.10197. PMID12655545. S2CID23769500.
Further reading
Dutta S, Goodsell D (January 2005). "Phenylalanine Hydroxylase". Molecule of the Month. RCSB Protein Data Bank (PDB). Archived from the original on 22 February 2018.
Regier DS, Greene CL (5 January 2017). "Phenylalanine Hydroxylase Deficiency". In Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, Gripp KW, Amemiya A (eds.). GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle. PMID20301677.
1dmw: CRYSTAL STRUCTURE OF DOUBLE TRUNCATED HUMAN PHENYLALANINE HYDROXYLASE WITH BOUND 7,8-DIHYDRO-L-BIOPTERIN
1j8t: Catalytic Domain of Human Phenylalanine Hydroxylase Fe(II)
1j8u: Catalytic Domain of Human Phenylalanine Hydroxylase Fe(II) in Complex with Tetrahydrobiopterin
1kw0: Catalytic Domain of Human Phenylalanine Hydroxylase (Fe(II)) in Complex with Tetrahydrobiopterin and Thienylalanine
1lrm: Crystal structure of binary complex of the catalytic domain of human phenylalanine hydroxylase with dihydrobiopterin (BH2)
1mmk: Crystal structure of ternary complex of the catalytic domain of human phenylalanine hydroxylase ((FeII)) complexed with tetrahydrobiopterin and thienylalanine
1mmt: Crystal structure of ternary complex of the catalytic domain of human phenylalanine hydroxylase (Fe(II)) complexed with tetrahydrobiopterin and norleucine
1pah: HUMAN PHENYLALANINE HYDROXYLASE DIMER, RESIDUES 117-424
1phz: STRUCTURE OF PHOSPHORYLATED PHENYLALANINE HYDROXYLASE
1tdw: Crystal structure of double truncated human phenylalanine hydroxylase BH4-responsive PKU mutant A313T.
1tg2: Crystal structure of phenylalanine hydroxylase A313T mutant with 7,8-dihydrobiopterin bound
2pah: TETRAMERIC HUMAN PHENYLALANINE HYDROXYLASE
2phm: STRUCTURE OF PHENYLALANINE HYDROXYLASE DEPHOSPHORYLATED
3pah: HUMAN PHENYLALANINE HYDROXYLASE CATALYTIC DOMAIN DIMER WITH BOUND ADRENALINE INHIBITOR
4pah: HUMAN PHENYLALANINE HYDROXYLASE CATALYTIC DOMAIN DIMER WITH BOUND NOR-ADRENALINE INHIBITOR
5pah: HUMAN PHENYLALANINE HYDROXYLASE CATALYTIC DOMAIN DIMER WITH BOUND DOPAMINE INHIBITOR
6pah: HUMAN PHENYLALANINE HYDROXYLASE CATALYTIC DOMAIN DIMER WITH BOUND L-DOPA (3,4-DIHYDROXYPHENYLALANINE) INHIBITOR
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.