Reaction catalysed
The enzyme converts lysine in proteins into hydroxylysine: [3]
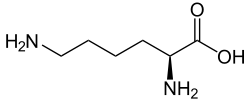




The reaction is important as a first step in cross-lnking collagen. [1]
| procollagen-lysine 1, 2-oxoglutarate 5-dioxygenase 1 | |||||||
|---|---|---|---|---|---|---|---|
| Identifiers | |||||||
| Symbol | PLOD1 | ||||||
| Alt. symbols | LLH, PLOD | ||||||
| NCBI gene | 5351 | ||||||
| HGNC | 9081 | ||||||
| OMIM | 153454 | ||||||
| RefSeq | NM_000302 | ||||||
| UniProt | Q02809 | ||||||
| Other data | |||||||
| EC number | 1.14.11.4 | ||||||
| Locus | Chr. 1 p36.3-36.2 | ||||||
| |||||||
| procollagen-lysine, 2-oxoglutarate 5-dioxygenase 2 | |||||||
|---|---|---|---|---|---|---|---|
| Identifiers | |||||||
| Symbol | PLOD2 | ||||||
| NCBI gene | 5352 | ||||||
| HGNC | 9082 | ||||||
| OMIM | 601865 | ||||||
| RefSeq | NM_000935 | ||||||
| UniProt | O00469 | ||||||
| Other data | |||||||
| Locus | Chr. 3 q24 | ||||||
| |||||||
Lysyl hydroxylases (or procollagen-lysine 5-dioxygenases) are alpha-ketoglutarate-dependent hydroxylases enzymes that catalyze the hydroxylation of lysine to hydroxylysine. [1] [2] Lysyl hydroxylases require iron and vitamin C as cofactors for their oxidation activity. It takes place (as a post-translational modification) following collagen synthesis in the cisternae (lumen) of the rough endoplasmic reticulum (ER). There are three lysyl hydroxylases (LH1-3) encoded in the human genome, namely: PLOD1, PLOD2 and PLOD3. From PLOD2 two splice variant can be expressed (LH2a and LH2b), where LH2b differs from LH2a by incorporating the small exon 13A. LH1 and LH3 hydroxylate lysyl residues in the collagen triple helix, whereas LH2b hydroxylates lysyl residues in the telopeptides of collagen. In addition to its hydroxylation activity, LH3 has glucosylation activity that produces disaccharide (Glc-Gal) attached to collagen hydroxylysines.
Collagen lysyl hydroxylation is the first step in collagen pyridinoline cross-linking, that is necessary for the stabilization of collagen.
The enzyme converts lysine in proteins into hydroxylysine: [3]
The reaction is important as a first step in cross-lnking collagen. [1]
Mutations in the PLOD1 gene have been linked to kyphoscoliotic Ehlers–Danlos syndrome (kEDS, in the past EDS VI). [4]
Mutations in the PLOD2 gene have been linked to Bruck syndrome in humans.
A deficiency in its cofactor vitamin C is associated with scurvy.
| | This oxidoreductase article is a stub. You can help Wikipedia by adding missing information. |