In humans, the enzyme is localized in endoplasmic reticulum membranes of cells in adrenal cortex,[14][15] and is encoded by the CYP21A2gene which is located near the CYP21A1Ppseudogene that has high degree of sequence similarity. This similarity makes it difficult to analyze the gene at the molecular level, and sometimes leads to loss-of-function mutations of the gene due to intergenic exchange of DNA.
Steroid 21-hydroxylase in humans is encoded by the CYP21A2 gene that may be accompanied by one or several copies of the nonfunctional pseudogeneCYP21A1P,[20][21] this pseudogene shares 98% of the exonic informational identity with the actual functional gene.[22][23]
Pseudogenes are common in genomes, and they originate as artifacts during the duplication process. Though often thought of as "junk DNA", research has shown that retaining these faulty copies can have a beneficial role, often providing regulation of their parent genes.[24]
In the mousegenome, the Cyp21a2 is a pseudogene and the Cyp21a1 is a functional gene.[25] In the chicken and quail, there is only a single Cyp21 gene, which locus is located between complement component C4 and TNX gene, along with Cenpa.[26]
Inside the MHC class III, CYP21A2 is located within the RCCX cluster (an abbreviation composed of the names of the genes RP (a former name for STK19 serine/threonine kinase 19),[30][31]C4, CYP21 and TNX),[32] which is the most complex gene cluster in the human genome.[33] The number of RCCX segments varies between one and four in a chromosome,[30] with the prevalence of approximately 15% for monomodular, 75% for bimodular (STK19-C4A-CYP21A1P-TNXA-STK19B-C4B-CYP21A2-TNXB),[31][34] and 10% for trimodular in Europeans.[35] The quadrimodular structure of the RCCX unit is very rare.[36][30][35] In a monomodular structure, all of the genes are functional i.e. protein-coding, but if a module count is two or more, there is only one copy of each functional gene rest being non-coding pseudogenes with the exception of the C4 gene which always has active copies.[30][35]
Due to the high degree of homology between the CYP21A2 gene and the CYP21A1P pseudogene and the complexity of the RCCX locus, it is difficult to perform molecular diagnostics for CYP21A2. The pseudogene can have single-nucleotide polymorphisms (SNP) that are identical or similar to those in the functional gene, making it difficult to distinguish between them. The pseudogene can also recombine with the functional gene, creating hybrid genes that have features of both. This can result in false-positive or false-negative results when testing for SNPs in the CYP21A2.[37]
The whole genome sequencing technology relies on breaking the DNA into small fragments, sequencing them, and then assembling them back together based on their overlaps. However, because of the high homology and variability of the CYP21A2 and its pseudogene, the fragments cannot be mapped unambiguously to either copy of the gene. This can lead to errors or gaps in the assembly, or missing some variants that are present in the gene.[38][37]
Polymerase chain reaction (PCR) molecular diagnostics uses selective primers to amplify specific segments of the DNA sequence that are relevant for diagnosing or detecting a certain disease or condition. If the primers are not designed carefully, they may bind to both the CYP21A2 and the CYP21A1P pseudogene, or to different segments of the RCCX cluster, resulting in false-positive or false-negative results. Therefore, PCR for the CYP21A2 requires the use of locus-specific primers that can distinguish between the gene and the pseudogene, and between different RCCX modules. Moreover, PCR may not be able to detect complex variants such as large gene conversions, deletions, or duplications, which are frequent in the case of the CYP21A2.[39][40][38]
Southern blotting, a method used for detecting and quantifying a specific DNA sequence in DNA samples, also has limitations in analyzing CYP21A2. This method is time-consuming and requires a large amount of good-quality DNA, which makes it less applicable in routine diagnostic settings. This method comes with a radioactive biohazard, which poses safety concerns and makes it labor-intensive. Southern blotting is unable to detect the junction sites of chimeras. The CYP21A2 gene is prone to mismatch and rearrangement, producing different types of complex variations that include copy number variants, large gene conversions, small insertions/deletions, and single-nucleotide (SNP) variants. Southern blotting is not capable of detecting all these types of variants simultaneously. Besides that, southern blotting requires genetic analysis of the parents, which is not always feasible or practical.[38][41]
Therefore, to analyze the CYP21A2 gene accurately, a more specialized and sensitive method is needed, such as targeted long-read sequencing, which can sequence longer DNA fragments and capture more information about the gene structure and variation. However, this method is not widely available or affordable for clinical use.[42][43][44]
Protein
Steroid 21-hydroxylase, is a member of the cytochrome P450 family of monooxygenase enzymes, the protein has 494 amino acid residues with a molecular weight of 55,000. This enzyme is at most 28% homologous to other P-450 enzymes that have been studied.[45]
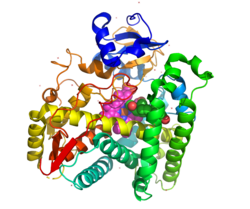
Structurally, the protein contains an evolutionarily conserved core of four α-helix bundles (the importance of such genetic conservation is in demonstrating the functional importance of this aspect of this protein's structure). In addition, it has two additional alpha helices, two sets of β-sheets, and a hemecofactor binding loop.[46] Each subunit in the human enzyme consists of a total of 13 α-helices and 9 β-strands that folds into a triangular prism-like tertiary structure.[12]
The iron(III) heme group that defines the active site resides in the center of each subunit. The human enzyme binds one substrate at a time.[12] In contrast, the well-characterized bovine enzyme can bind two substrates.[47] The human and bovine enzyme share 80% amino acid sequence identity, but are structurally different, particularly in loop regions, and also evident in secondary structure elements.[12]
Species
Variations of the steroid 21-hydroxylase can be found in all vertebrates.[48]
Cyp21 first emerged in chordates before the speciation between basal chordates and vertebrates.[49] The sea lamprey, an early jawless fish species that originated over 500 million years ago, provides valuable insights into the evolution and emergence of Cyp21. Sea lampreys lack the 11β-hydroxylase enzyme responsible for converting 11-deoxycortisol to cortisol as observed in mammals. Instead, they rely on 11-deoxycortisol, a product of a reaction catalyzed by CYP21, as their primary glucocorticoid hormone with mineralocorticoid properties. This suggests the presence of a complex and highly specific corticosteroid signaling pathway that emerged at least half a billion years ago during early vertebrate evolution.[50]
In vertebrates, such as fish, amphibians, reptiles, birds, and mammals, Cyp21 participates in the biosynthesis of glucocorticoids and mineralocorticoids, therefore, Cyp21 is essential for the regulation of stress response, electrolyte balance and blood pressure, immune system, and metabolism in vertebrates.[51]
Cyp21 is relatively conserved among mammals, and shows some variations in its structure, expression, and regulation.[51] Rhesus macaques and orangutans possess two copies of Cyp21, while chimpanzees have three, still, a pseudogene (CYP21A1P) is only present in humans among primates.[52]
Unlike other enzymes of the cytochrome P450 superfamily of enzymes that are expressed in multiple tissues, with most abundant expression in the liver, in adult humans steroid 21-hydroxylase, along with steroid 11β-hydroxylase and aldosterone synthase, is almost exclusively expressed in the adrenal gland.[54][55]
As of 2023,[update] the main subcellular location for the encoded protein in human cells is not known, and is pending cell analysis.[56]
Function
Steroid numbering, C21 is in the side chain of C17
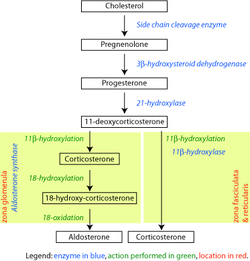
The enzyme, steroid 21-hydroxylase hydroxylatessteroids at the C21 position.[13] Steroids are a group of naturally occurring and synthetically produced organic compounds, steroids all share a four ring primary structure. The enzyme catalyzes the chemical reaction in which the hydroxyl group (-OH) is added at the C21 position of the steroid biomolecule. This location is on a side chain of the D ring.
The enzyme is a member of the cytochrome P450 superfamily of monooxygenase enzymes. The cytochrome P450 enzymes catalyze many reactions involved in drug metabolism and synthesis of cholesterol, steroids and other lipids.
Like other cytochrome P450 enzymes, steroid 21-hydroxylase participates in the cytochrome P450 catalytic cycle and engages in one-electron transfer with NADPH-P450 reductase. Steroid 21-hydroxylase is highly specific for hydroxylation of progesterone and 17-hydroxyprogesterone. This is in marked contrast to the evolutionarily and functionally related P450 enzyme 17-hydroxylase, which has a broad range of substrates.[59]
Studies of the human enzyme expressed in yeast initially classified 17-hydroxyprogesterone as the preferred substrate for steroid 21-hydroxylase,[59][62][63] however, later analysis of the purified human enzyme found a lower KM and greater catalytic efficiency for progesterone over 17-hydroxyprogesterone.[12]
The catalytic efficiency of steroid 21-hydroxylase for conversion of progesterone in humans is approximately 1.3 x 107 M−1s−1 at 37°C. This makes it the most catalytically efficient P450 enzyme of those reported to date, and catalytically more efficient than the closely related bovine steroid 21-hydroxylase enzyme.[14] C-H bond breaking to create a primary carbon radical is thought to be the rate-limiting step in the hydroxylation.[12]
Genetic variants in the CYP21A2 gene cause a disturbance in the development of the enzyme, leading to congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency. Gene conversion events involving the functional gene and the pseudogene account for many cases of steroid 21-hydroxylase deficiency.[64] CAH is an autosomal recessive disorder. There are multiple forms of CAH, defined as classical and nonclassical forms based on the amount of enzyme function still present in the patient.
The classical forms occur in approximately 1 in 10000 to 1 in 20000 births globally,[58][65] and includes both the salt-wasting (excessive excretion of sodium via the urine causing hyponatremia and dehydration) and simple-virilizing forms. Complete loss of enzymatic activity causes the salt-wasting form. Variations in the structure of steroid 21-hydroxylase are related to the clinical severity of congenital adrenal hyperplasia. Cortisol and aldosterone deficits are associated with life-threatening sodium loss, as the steroids play roles in regulating sodiumhomeostasis. Simple-virilizing CAH patients (~1-2% enzyme function)[58] maintain adequate sodium homeostasis, but exhibit other symptoms shared by the salt-wasting form, including accelerated growth in childhood and ambiguous genitalia in female neonates.
The nonclassical form is the mildest condition, retaining about 20% to 50% of enzyme function.[58] This form is associated with mild and clinically silent cortisol impairment,[65] but an excess of androgens post-puberty.[66]
Non-classical congenital adrenal hyperplasia caused by 21-hydroxylase deficiency (NCCAH) is a milder and late-onset congenital adrenal hyperplasia. Its prevalence rate in different ethnic groups varies from 1 in 1000 to 1 in 50.[58] Some people affected by the condition have no relevant signs and symptoms, while others experience symptoms of hyperandrogenism.[58][65][66]
Women with NCCAH usually have normal female genitalia at birth. In later life, the signs and symptoms of the condition may include acne, hirsutism, male-pattern baldness, irregular menstruation, and infertility.[58][65][25]
Fewer studies have been published about males with NCCAH comparing to those about females, because males are generally asymptomatic.[25][58] Males, however, may present with acne[67][68] and early balding.[69][70]
While symptoms are usually diagnosed after puberty, children may present with premature adrenarche.[71]
Research on other conditions
There is ongoing research on how Genetic variants in the CYP21A2 gene may lead to pathogenic conditions. A variant of this gene has been reported to cause autosomal dominant posterior polar cataract, suggesting that steroid 21-hydroxylase may be involved in the extra-adrenal biosynthesis of aldosterone and cortisol in the lens of the eye.[72]
History
In the 1950s and 1960s, steroidogenic pathways that included cholesterol conversion to progesterone through a complex pathway involving multiple steps were identified, and, among them, a pathway for cortisol synthesis showing specific enzymatic steps that included hydroxylation reactions at position 21 (21-hydroxylation) mediated by cytochrome P450 enzymes.[73] Cytochrome P450 enzymes were then described, and steroid 21-hydroxylation was associated with cytochrome P450.[74][73]
In the 1980s and 1990s, partial-length bovine Cyp21 cDNA clones were identified as related to human CYP21A2.[75][73] Researchers discovered mutations in the CYP21A2 gene associated with congenital adrenal hyperplasia (CAH).[73]
From the 1990s onward, specific mutations were correlated with different forms/severity levels of CAH. Genotype/phenotype correlations were investigated for improved diagnostic accuracy.[73]
↑ "UniProt". www.uniprot.org. Archived from the original on 28 November 2023. Retrieved 26 November 2023.
↑ Marino S, Perez Garrido N, Ramírez P, Pujana M, Dratler G, Belgorosky A, Marino R (2020). "Molecular analysis of the CYP21A2 gene in dried blood spot samples". Medicina. 80 (3): 197–202. PMID32442933.
↑ Kaewkot A, Boonkaewwan C, Noosud J, Kayan A (November 2017). "Expression level of the cytochrome P450c21 (CYP21) protein correlating to drip loss in pigs". Animal Science Journal. 88 (11): 1855–1859. doi:10.1111/asj.12863. PMID28677294.
↑ Sarafoglou K, Lorentz CP, Otten N, Oetting WS, Grebe SK (July 2012). "Molecular testing in congenital adrenal hyperplasia due to 21α-hydroxylase deficiency in the era of newborn screening". Clinical Genetics. 82 (1): 64–70. doi:10.1111/j.1399-0004.2011.01694.x. PMID21534945. S2CID7197547.
↑ Bergamaschi R, Livieri C, Uggetti C, Candeloro E, Egitto MG, Pichiecchio A, Cosi V, Bastianello S (March 2006). "Brain white matter impairment in congenital adrenal hyperplasia". Archives of Neurology. 63 (3): 413–6. doi:10.1001/archneur.63.3.413. PMID16540460.
↑ Marcol W, Kalina-Faska B, Wackermann-Ramos A, Koehler B (2000). "Congenital adrenal hyperplasia conditioned by 21beta-hydroxylase deficiency - clinical considerations". Endokrynologia, Diabetologia I Choroby Przemiany Materii Wieku Rozwojowego (in Polish). 6 (1): 67–9. PMID14640134.
1 2 This article incorporates public domain material from "NCBI: CYP21A2 cytochrome P450 family 21 subfamily A member 2". Reference Sequence collection. National Center for Biotechnology Information. Retrieved 30 November 2020. This gene encodes a member of the cytochrome P450 superfamily of enzymes. The cytochrome P450 proteins are monooxygenases that catalyze many reactions involved in drug metabolism and synthesis of cholesterol, steroids, and other lipids. This protein localizes to the endoplasmic reticulum and hydroxylates steroids at the 21 position. Its activity is required for the synthesis of steroid hormones including cortisol and aldosterone. Mutations in this gene cause congenital adrenal hyperplasia. A related pseudogene is located near this gene; gene conversion events involving the functional gene and the pseudogene are thought to account for many cases of steroid 21-hydroxylase deficiency. Two transcript variants encoding different isoforms have been found for this gene.
1 2 Neunzig J, Milhim M, Schiffer L, Khatri Y, Zapp J, Sánchez-Guijo A, etal. (March 2017). "The steroid metabolite 16(β)-OH-androstenedione generated by CYP21A2 serves as a substrate for CYP19A1". The Journal of Steroid Biochemistry and Molecular Biology. 167: 182–191. doi:10.1016/j.jsbmb.2017.01.002. PMID28065637. S2CID36860068.
1 2 3 Karaoğlan M, Nacarkahya G, Aytaç EH, Keskin M (November 2021). "Challenges of CYP21A2 genotyping in children with 21-hydroxylase deficiency: determination of genotype-phenotype correlation using next generation sequencing in Southeastern Anatolia". J Endocrinol Invest. 44 (11): 2395–2405. doi:10.1007/s40618-021-01546-z. PMID33677812. S2CID232133292.
↑ Zhang X, Gao Y, Lu L, Cao Y, Zhang W, Sun B, Wu X, Tong A, Chen S, Wang X, Mao J, Nie M (2023). "Targeted long-read sequencing for comprehensive detection of CYP21A2 mutations in patients with 21-hydroxylase deficiency". Journal of Endocrinological Investigation. 47 (4): 833–841. doi:10.1007/s40618-023-02197-y. PMID37815751. S2CID263800944.
↑ Baker ME, Nelson DR, Studer RA (July 2015). "Origin of the response to adrenal and sex steroids: Roles of promiscuity and co-evolution of enzymes and steroid receptors". J Steroid Biochem Mol Biol. 151: 12–24. doi:10.1016/j.jsbmb.2014.10.020. PMID25445914. S2CID21649057.
1 2 Cameron PU, Tabarias HA, Pulendran B, Robinson W, Dawkins RL (1990). "Conservation of the central MHC genome: PFGE mapping and RFLP analysis of complement, HSP70, and TNF genes in the goat". Immunogenetics. 31 (4): 253–64. doi:10.1007/BF00204897. PMID1970334. S2CID22716959.
↑ Araújo RS, Mendonca BB, Barbosa AS, Lin CJ, Marcondes JA, Billerbeck AE, Bachega TA (October 2007). "Microconversion between CYP21A2 and CYP21A1P promoter regions causes the nonclassical form of 21-hydroxylase deficiency". The Journal of Clinical Endocrinology and Metabolism. 92 (10): 4028–34. doi:10.1210/jc.2006-2163. PMID17666484.
1 2 Auchus RJ, Sampath Kumar A, Andrew Boswell C, Gupta MK, Bruce K, Rath NP, Covey DF (January 2003). "The enantiomer of progesterone (ent-progesterone) is a competitive inhibitor of human cytochromes P450c17 and P450c21". Archives of Biochemistry and Biophysics. 409 (1): 134–44. doi:10.1016/s0003-9861(02)00491-5. PMID12464252.
↑ Rosenfeld G, Ungar F, Dorfman RI, Pincus G (1955). "Irradiation and adrenal steroidogenesis: steroid transformations by irradiated isolated perfused calf adrenals". Endocrinology. 56 (1): 24–9. doi:10.1210/endo-56-1-24. PMID13220521.
↑ Dorfman RI, Hayano M (March 1952). "The action of adrenal homogenates on progesterone, 17-hydroxyprogesterone and 21-desoxycortisone". Archives of Biochemistry and Biophysics. 36 (1): 237–9. doi:10.1016/0003-9861(52)90397-4. PMID14934270.
↑ Lorence MC, Trant JM, Mason JI, Bhasker CR, Fujii-Kuriyama Y, Estabrook RW, Waterman MR (August 1989). "Expression of a full-length cDNA encoding bovine adrenal cytochrome P450C21". Archives of Biochemistry and Biophysics. 273 (1): 79–88. doi:10.1016/0003-9861(89)90164-1. PMID2502949.
↑ Wu DA, Hu MC, Chung BC (April 1991). "Expression and functional study of wild-type and mutant human cytochrome P450c21 in Saccharomyces cerevisiae". DNA and Cell Biology. 10 (3): 201–9. doi:10.1089/dna.1991.10.201. PMID1707279.
↑ "NCBI: CYP21A2 cytochrome P450 family 21 subfamily A member 2". National Center for Biotechnology Information. Archived from the original on 28 October 2020. Retrieved 30 November 2020. This gene encodes a member of the cytochrome P450 superfamily of enzymes. The cytochrome P450 proteins are monooxygenases that catalyze many reactions involved in drug metabolism and synthesis of cholesterol, steroids, and other lipids. This protein localizes to the endoplasmic reticulum and hydroxylates steroids at the 21 position. Its activity is required for the synthesis of steroid hormones including cortisol and aldosterone. Mutations in this gene cause congenital adrenal hyperplasia. A related pseudogene is located near this gene; gene conversion events involving the functional gene and the pseudogene are thought to account for many cases of steroid 21-hydroxylase deficiency. Two transcript variants encoding different isoforms have been found for this gene. This article incorporates text from this source, which is in the public domain.
↑ Sharquie KE, Noaimi AA, Saleh BO, Anbar ZN (December 2009). "The frequency of 21-alpha hydroxylase enzyme deficiency and related sex hormones in Iraqi healthy male subjects versus patients with acne vulgaris". Saudi Medical Journal. 30 (12): 1547–50. PMID19936418.
↑ Falhammar H, Nordenström A (September 2015). "Nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency: clinical presentation, diagnosis, treatment, and outcome". Endocrine. 50 (1): 32–50. doi:10.1007/s12020-015-0656-0. PMID26082286. S2CID23469344.
↑ Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dungan K, Grossman A, Hershman JM, Hofland J, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Purnell J, Singer F, Stratakis CA, Trence DL, Wilson DP, Yau M, Gujral J, New MI (April 2019). Congenital Adrenal Hyperplasia: Diagnosis and Emergency Treatment. MDText.com. PMID25905311. Archived from the original on 14 November 2020. Retrieved 25 March 2021.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.











{kind=link}
{kind=link}