
Collagen is the main structural protein in the extracellular matrix found in the body's various connective tissues. As the main component of connective tissue, it is the most abundant protein in mammals, making up from 25% to 35% of the whole-body protein content. Collagen consists of amino acids bound together to form a triple helix of elongated fibril known as a collagen helix. It is mostly found in connective tissue such as cartilage, bones, tendons, ligaments, and skin. Vitamin C is vital for collagen synthesis, and Vitamin E improves the production of collagen.

Osteogenesis imperfecta, colloquially known as brittle bone disease, is a group of genetic disorders that all result in bones that break easily. The range of symptoms—on the skeleton as well as on the body's other organs—may be mild to severe. Symptoms found in various types of OI include whites of the eye (sclerae) that are blue instead, short stature, loose joints, hearing loss, breathing problems and problems with the teeth. Potentially life-threatening complications, all of which become more common in more severe OI, include: tearing (dissection) of the major arteries, such as the aorta; pulmonary valve insufficiency secondary to distortion of the ribcage; and basilar invagination.

Collagen, type II, alpha 1 , also known as COL2A1, is a human gene that provides instructions for the production of the pro-alpha1(II) chain of type II collagen.
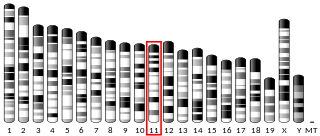
A disintegrin and metalloproteinase with thrombospondin motifs 2 (ADAM-TS2) also known as procollagen I N-proteinase is an enzyme that in humans is encoded by the ADAMTS2 gene.

Collagen, type I, alpha 1, also known as alpha-1 type I collagen, is a protein that in humans is encoded by the COL1A1 gene. COL1A1 encodes the major component of type I collagen, the fibrillar collagen found in most connective tissues, including cartilage.

Collagen alpha-2(XI) chain is a protein that in humans is encoded by the COL11A2 gene.

Dentinogenesis imperfecta (DI) is a genetic disorder of tooth development. It is inherited in an autosomal dominant pattern, as a result of mutations on chromosome 4q21, in the dentine sialophosphoprotein gene (DSPP). It is one of the most frequently occurring autosomal dominant features in humans. Dentinogenesis imperfecta affects an estimated 1 in 6,000-8,000 people.
Lysyl hydroxylases are alpha-ketoglutarate-dependent hydroxylases enzymes that catalyze the hydroxylation of lysine to hydroxylysine. Lysyl hydroxylases require iron and vitamin C as cofactors for their oxidation activity. It takes place following collagen synthesis in the cisternae (lumen) of the rough endoplasmic reticulum (ER). There are three lysyl hydroxylases (LH1-3) encoded in the human genome, namely: PLOD1, PLOD2 and PLOD3. From PLOD2 two splice variant can be expressed, where LH2b differs from LH2a by incorporating the small exon 13A. LH1 and LH3 hydroxylate lysyl residues in the collagen triple helix, whereas LH2b hydroxylates lysyl residues in the telopeptides of collagen. In addition to its hydroxylation activity, LH3 has glycosylation activity that produces either monosaccharide (Gal) or disaccharide (Glc-Gal) attached to collagen hydroxylysines.

Sack–Barabas syndrome is an older name for the medical condition vascular Ehlers–Danlos syndrome (vEDS). It affects the body's blood vessels and organs, making them prone to rupture.
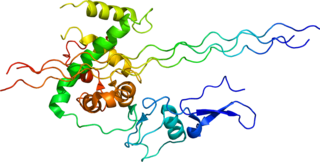
Type III Collagen is a homotrimer, or a protein composed of three identical peptide chains (monomers), each called an alpha 1 chain of type III collagen. Formally, the monomers are called collagen type III, alpha-1 chain and in humans are encoded by the COL3A1 gene. Type III collagen is one of the fibrillar collagens whose proteins have a long, inflexible, triple-helical domain.
Type V collagen is a form of fibrillar collagen associated with classical Ehlers-Danlos syndrome. It is found within the dermal/epidermal junction, placental tissues, as well as in association with tissues containing type I collagen.

Collagen alpha-1(V) chain is a protein that in humans is encoded by the COL5A1 gene.

Collagen alpha-2(V) chain is a protein that in humans is encoded by the COL5A2 gene.

Collagen alpha-2(I) chain is a protein that in humans is encoded by the COL1A2 gene.

Collagen alpha-1(XI) chain is a protein that in humans is encoded by the COL11A1 gene.

Collagen alpha-1(VIII) chain is a protein that in humans is encoded by the COL8A1 gene.

Collagen alpha-3(V) chain is a protein that in humans is encoded by the COL5A3 gene.

Carbohydrate sulfotransferase 14 is an enzyme that in humans is encoded by the CHST14 gene.

Bruck syndrome is characterized as the combination of arthrogryposis multiplex congenita and osteogenesis imperfecta. Both diseases are uncommon, but concurrence is extremely rare which makes Bruck syndrome very difficult to research. Bruck syndrome is thought to be an atypical variant of osteogenesis imperfecta most resembling type III, if not its own disease. Multiple gene mutations associated with osteogenesis imperfecta are not seen in Bruck syndrome. Many affected individuals are within the same family, and pedigree data supports that the disease is acquired through autosomal recessive inheritance. Bruck syndrome has features of congenital contractures, bone fragility, recurring bone fractures, flexion joint and limb deformities, pterygia, short body height, and progressive kyphoscoliosis. Individuals encounter restricted mobility and pulmonary function. A reduction in bone mineral content and larger hydroxyapatite crystals are also detectable Joint contractures are primarily bilateral and symmetrical, and most prone to ankles. Bruck syndrome has no effect on intelligence, vision, or hearing.

Daniel S. Greenspan is an American biomedical scientist, academic and researcher. He is Kellett professor of Cell and Regenerative Biology at the University of Wisconsin-Madison School of Medicine and Public Health. He has authored over 120 publications. His research has mainly focused on genes encoding proteins of the extracellular space and possible links between defects in such genes and human development and disease.
