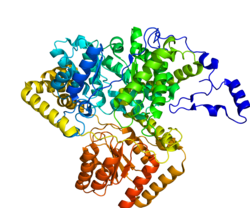
Methylmalonyl-CoA mutase (EC5.4.99.2, MCM), mitochondrial, also known as methylmalonyl-CoA isomerase, is a protein that in humans is encoded by the MUT gene. This vitamin B12-dependent enzyme catalyzes the isomerization of methylmalonyl-CoA to succinyl-CoA in humans. Mutations in MUT gene may lead to various types of methylmalonic aciduria.[5]
MCM was first identified in ratliver and sheepkidney in 1955. In its latent form, it is 750 amino acids in length. Upon entry to the mitochondria, the 32 amino acid mitochondrial leader sequence at the N-terminus of the protein is cleaved, forming the fully processed monomer. The monomers then associate into homodimers, and bind AdoCbl (one for each monomer active site) to form the final, active holoenzyme form.[6]
Structure
Gene
The MUT gene lies on the chromosome location of 6p12.3 and consists of 13 exons, spanning over 35kb.[7]
Protein
The mature enzyme is a homodimer with the N-terminal CoA binding domain and the C- terminal cobalamin-binding domain.[8]
Function
Methylmalonyl-CoA mutase is expressed in high concentrations in the kidney, in intermediate concentrations in the heart, ovaries, brain, muscle, and liver, and in low concentrations in the spleen.[6] The enzyme can be found all throughout the central nervous system (CNS).[6] MCM resides in the mitochondria, where a number of substances, including the branched-chain amino acidsisoleucine and valine, as well as methionine, threonine, thymine and odd-chain fatty acids, are metabolized via methylmalonate semialdehyde (MMlSA) or propionyl-CoA (Pr-CoA) to a common compound - methylmalonyl-CoA (MMl-CoA). MCM catalyzes the reversible isomerisation of l‐methylmalonyl‐CoA to succinyl‐CoA, requiring cobalamin (vitamin B12) in the form of adenosylcobalamin (AdoCbl) as a cofactor. As an important step in propionate catabolism, this reaction is required for the degradation of odd-chain fatty acids, the amino acids valine, isoleucine, methionine, and threonine, and cholesterol,[9] funneling metabolites from the breakdown of these amino acids into the tricarboxylic acid cycle.[10]
Methylmalonyl-CoA mutase catalyzes the following reaction:
Either mutations to the gene MUT (encodes methylmalonyl-CoA mutase), or MMAA (encodes a chaperone protein of methylmalonyl-CoA mutase, MMAA protein) can lead to methylmalonyl acidemia.[12] Mutations to MUT can be categorized as either MUT0 (demonstrates no activity even in presence of excess AdoCbl), or MUT1 (demonstrates very low activity in presence of excess AdoCbl).[8] Over half of the mutations of MUT are missense mutations[10] while nonsense mutations comprise a significant remaining fraction (approximately 14%)[13]
Common treatment methods for MMA include a liver transplant or a liver and kidney transplant to combat the renal disease of methylmalonic acidemia. However, detrimental neurological effects can continue to plague patients even after a successful operation. It is thought that this is due to the widespread presence of methylmalonyl-CoA mutase throughout the central nervous system. Due to the loss of functionality of the enzyme, substrate levels build up in the CNS. The substrate, L-methylmalonyl-CoA hydrolyzes to form methylmalonate (methylmalonic acid), a neurotoxic dicarboxylic acid that, due to the poor dicarboxylic acid transport capacities of the blood-brain barrier, is effectively trapped within the CNS, leading to neurological debilitation. To combat these effects perioperative anti-catabolic regimes and no diet discontinuation are recommended.[6]
The murine model has proven an adequate and accurate way of studying the effects of MMA, and potential treatment methods.[14][15]
Mechanism
MCM's reaction mechanism
The MCM reaction mechanism begins with homolytic cleavage of AdoB12's C-Co(III) bond, the C and Co atoms each acquire one of the electrons that formed the cleaved electron pair bond. The Co ion, therefore, fluctuates between its Co(III) and Co(II) oxidation states [the two states are spectroscopically distinguishable: Co(III) is red and diamagnetic (no unpaired electrons), whereas Co(II) is yellow and paramagnetic (unpaired electrons)]. Hence, the role of coenzyme B-12 in the catalytic process is that of a reversible generator of a free radical. The C-Co(III) bond is weak, with a dissociation energy = 109 kJ/mol, and appears to be further weakened through steric interactions with the enzyme. The homolytic reaction is unusual in biology, as is the presence of a metal-carbon bond.
Methylmalonyl-CoA mutase is a member of the isomerase subfamily of adenosylcobalamin-dependent enzymes. Furthermore, it is classified as class I, as it is a ‘DMB-off’/’His-on’ enzyme. This refers to the nature of the AdoCbl cofactor in the active site of methylmalonyl CoA.[16] AdoCbl is composed of a central cobalt-containing corrin ring, an upper axial ligand (β-axial ligand), and a lower axial ligand (α-axial ligand). In methylmalonyl-CoA mutase, the β-axial ligand 5’-deoxy-5’-adenosine reversibly dissociated to give the deoxyadenosyl radical. The α-axial ligand 5,6-dimethylbenzimidazole (DMB) is involved in organizing the active site to enable histidine-610 to bond with Co, instead of DMB (the reason for the ‘DMB-off’/’His-on’ notation).[16] Binding of histidine-610 residue increases the rate of homolytic β-axial ligand – Co bond breakage by a factor of 1012.[17]
MCM active site. Corrin ring and α-axial ligand (DMB): (yellow), β-axial ligand: (green), substrate/product: (cyan), residues interacting with β-axial ligand: glu370, asn366, gly91, ala139 (blue), residues interacting with substrate: gln197, his244, arg207, tyr89 (red), and his610: (orange). Rendered from PDB 4REQ.
Other important residues of methylmalonyl-CoA mutase include Histidine-244, which acts as a general acid near the substrate and shields the radical species from side reactions involving oxygen,[20]Glutamate-370, whose hydrogen bond with the 2’-OH group of the ribose of the β-axial ligand forces interaction between the β-axial ligand radical species and the substrate,[21] and tyrosine-89 which stabilizes reactive radical intermediates and accounts for the stereo-selectivity of the enzyme.[18][22]
The processing protein, MMAA protein, fills the important role of aiding cofactor loading and exchange.[12][23]MMAA protein favors association with the MCM apoenzyme, and allows for the transfer of the AdoCbl cofactor to the enzyme active site.[23] Furthermore, if the bound AdoCbl accrues oxidative damage during normal functioning, MMAA protein fosters exchange of the damaged cofactor for a new AdoCbl via a GTP-reliant pathway.[12][23]
↑ "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
↑ "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
1 2 Keyfi F, Sankian M, Moghaddassian M, Rolfs A, Varasteh AR (January 2016). "Molecular, biochemical, and structural analysis of a novel mutation in patients with methylmalonyl-CoA mutase deficiency". Gene. 576 (1 Pt 2): 208–13. doi:10.1016/j.gene.2015.10.002. PMID26449400.
1 2 3 4 Ballhausen D, Mittaz L, Boulat O, Bonafé L, Braissant O (December 2009). "Evidence for catabolic pathway of propionate metabolism in CNS: expression pattern of methylmalonyl-CoA mutase and propionyl-CoA carboxylase alpha-subunit in developing and adult rat brain". Neuroscience. 164 (2): 578–87. doi:10.1016/j.neuroscience.2009.08.028. PMID19699272. S2CID34612963.
↑ Dündar H, Özgül RK, Güzel-Ozantürk A, Dursun A, Sivri S, Aliefendioğlu D, Coşkun T, Tokatli A (August 2012). "Microarray based mutational analysis of patients with methylmalonic acidemia: identification of 10 novel mutations". Molecular Genetics and Metabolism. 106 (4): 419–23. doi:10.1016/j.ymgme.2012.05.014. PMID22727635.
1 2 3 Takahashi-Íñiguez T, García-Arellano H, Trujillo-Roldán MA, Flores ME (January 2011). "Protection and reactivation of human methylmalonyl-CoA mutase by MMAA protein". Biochemical and Biophysical Research Communications. 404 (1): 443–7. Bibcode:2011BBRC..404..443T. doi:10.1016/j.bbrc.2010.11.141. PMID21138732.
↑ Buck NE, Wood LR, Hamilton NJ, Bennett MJ, Peters HL (November 2012). "Treatment of a methylmalonyl-CoA mutase stopcodon mutation". Biochemical and Biophysical Research Communications. 427 (4): 753–7. Bibcode:2012BBRC..427..753B. doi:10.1016/j.bbrc.2012.09.133. PMID23041189.
↑ Buckel W, Friedrich P, Golding BT (October 2012). "Hydrogen bonds guide the short-lived 5'-deoxyadenosyl radical to the place of action". Angewandte Chemie. 51 (40): 9974–6. doi:10.1002/anie.201205299. PMID22945861.
↑ Thomä NH, Meier TW, Evans PR, Leadlay PF (October 1998). "Stabilization of radical intermediates by an active-site tyrosine residue in methylmalonyl-CoA mutase". Biochemistry. 37 (41): 14386–93. CiteSeerX10.1.1.608.304. doi:10.1021/bi981375o. PMID9772164.
Lubrano R, Elli M, Rossi M, Travasso E, Raggi C, Barsotti P, Carducci C, Berloco P (August 2007). "Renal transplant in methylmalonic acidemia: could it be the best option? Report on a case at 10 years and review of the literature". Pediatric Nephrology. 22 (8): 1209–14. doi:10.1007/s00467-007-0460-z. PMID17401587. S2CID24610554.
Frenkel EP, Kitchens RL (December 1975). "Intracellular localization of hepatic propionyl-CoA carboxylase and methylmalonyl-CoA mutase in humans and normal and vitamin B12 deficient rats". British Journal of Haematology. 31 (4): 501–13. doi:10.1111/j.1365-2141.1975.tb00885.x. PMID24458. S2CID1232083.
Crane AM, Martin LS, Valle D, Ledley FD (May 1992). "Phenotype of disease in three patients with identical mutations in methylmalonyl CoA mutase". Human Genetics. 89 (3): 259–64. doi:10.1007/BF00220536. PMID1351030. S2CID5624280.
Zoghbi HY, O'Brien WE, Ledley FD (November 1988). "Linkage relationships of the human methylmalonyl CoA mutase to the HLA and D6S4 loci on chromosome 6". Genomics. 3 (4): 396–8. doi:10.1016/0888-7543(88)90135-8. PMID2907507.
Fenton WA, Hack AM, Willard HF, Gertler A, Rosenberg LE (April 1982). "Purification and properties of methylmalonyl coenzyme A mutase from human liver". Archives of Biochemistry and Biophysics. 214 (2): 815–23. doi:10.1016/0003-9861(82)90088-1. PMID6124211.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.