The western blot (sometimes called the protein immunoblot), or western blotting, is a widely used analytical technique in molecular biology and immunogenetics to detect specific proteins in a sample of tissue homogenate or extract,[1] and to visualize, distinguish, and quantify the different proteins in a complicated protein combination.[2]
Western blot technique uses three elements to achieve its task of separating a specific protein from a complex: separation by size, transfer of protein to a solid support, and marking target protein using a primary and secondary antibody to visualize.[1] A synthetic or animal-derivedantibody (known as the primary antibody) is created that recognizes and binds to a specific target protein. The electrophoresis membrane is washed in a solution containing the primary antibody, before excess antibody is washed off.[3] A secondary antibody is added which recognizes and binds to the primary antibody. The secondary antibody is visualized through various methods such as staining, immunofluorescence, and radioactivity, allowing indirect detection of the specific target protein.[3]
The name western blot is a play on the Southern blot, a technique for DNA detection named after its inventor, English biologist Edwin Southern. Similarly, detection of RNA is termed as northern blot.[4] The term western blot was given by W. Neal Burnette in 1981,[5] although the method, but not the name, was independently invented in 1979 by Jaime Renart, Jakob Reiser, and George Stark,[6] and by Harry Towbin, Theophil Staehelin, and Julian Gordon at the Friedrich Miescher Institute in Basel, Switzerland.[7] The Towbin group also used secondary antibodies for detection, thus resembling the actual method that is almost universally used today. Between 1979 and 2019 "it has been mentioned in the titles, abstracts, and keywords of more than 400,000 PubMed-listed publications" and may still be the most-used protein-analytical technique.[8]
Applications
Western blot HIV test where the first two strips are negative and positive controls followed by actual tests
The western blot is extensively used in biochemistry for the qualitative detection of single proteins and protein-modifications (such as post-translational modifications). At least 8–9% of all protein-related publications are estimated to apply western blots.[8] It is used as a general method to identify the presence of a specific single protein within a complex mixture of proteins. A semi-quantitative estimation of a protein can be derived from the size and colour intensity of a protein band on the blot membrane. In addition, applying a dilution series of a purified protein of known concentrations can be used to allow a more precise estimate of protein concentration. The western blot is routinely used for verification of protein production after cloning. It is also used in medical diagnostics, e.g., in the HIV test or BSE-Test.[9]
The confirmatory HIV test employs a western blot to detect anti-HIV antibody in a human serum sample. Proteins from known HIV-infected cells are separated and blotted on a membrane as above. Then, the serum to be tested is applied in the primary antibody incubation step; free antibody is washed away, and a secondary anti-human antibody linked to an enzyme signal is added. The stained bands then indicate the proteins to which the patient's serum contains antibody.[10] A western blot is also used as the definitive test for variant Creutzfeldt–Jakob disease, a type of prion disease linked to the consumption of contaminated beef from cattle with bovine spongiform encephalopathy (BSE, commonly referred to as 'mad cow disease').[11] Another application is in the diagnosis of tularemia. An evaluation of the western blot's ability to detect antibodies against F. tularensis revealed that its sensitivity is almost 100% and the specificity is 99.6%.[12] Some forms of Lyme disease testing employ western blotting.[13] A western blot can also be used as a confirmatory test for Hepatitis B infection and HSV-2 (Herpes Type 2) infection.[14][15] In veterinary medicine, a western blot is sometimes used to confirm FIV+ status in cats.[16]
Further applications of the western blot technique include its use by the World Anti-Doping Agency (WADA). Blood doping is the misuse of certain techniques and/or substances to increase one's red blood cell mass, which allows the body to transport more oxygen to muscles and therefore increase stamina and performance. There are three widely known substances or methods used for blood doping, namely, erythropoietin (EPO), synthetic oxygen carriers and blood transfusions. Each is prohibited under WADA's List of Prohibited Substances and Methods. The western blot technique was used during the 2014 FIFA World Cup in the anti-doping campaign for that event.[17] In total, over 1000 samples were collected and analysed by Reichel, et al.[18] in the WADA accredited Laboratory of Lausanne, Switzerland. Recent research utilizing the western blot technique showed an improved detection of EPO in blood and urine based on novel Velum SAR precast horizontal gels optimized for routine analysis.[19] With the adoption of the horizontal SAR-PAGE in combination with the precast film-supported Velum SAR gels the discriminatory capacity of micro-dose application of rEPO was significantly enhanced.
Identification of protein localization across cells
For medication development, the identification of therapeutic targets, and biological research, it is essential to comprehend where proteins are located within a cell.[2][20] The subcellular locations of proteins inside the cell and their functions are closely related. The relationship between protein function and localization suggests that when proteins move, their functions may change or acquire new characteristics. A protein's subcellular placement can be determined using a variety of methods. Numerous efficient and reliable computational tools and strategies have been created and used to identify protein subcellular localization.[21] With the aid of subcellular fractionation methods, WB continues to be an important fundamental method for the investigation and comprehension of protein localization.[2]
Epitope mapping
Due to their various epitopes, antibodies have gained interest in both basic and clinical research. The foundation of antibody characterization and validation is epitope mapping. The procedure of identifying an antibody's binding sites (epitopes) on the target protein is referred to as "epitope mapping." Finding the binding epitope of an antibody is essential for the discovery and creation of novel vaccines, diagnostics, and therapeutics.[2] As a result, various methods for mapping antibody epitopes have been created. At this point, western blotting's specificity is the main feature that sets it apart from other epitope mapping techniques. There are several application of western blot for epitope mapping on human skin samples, hemorrhagic disease virus.[2][22][23]
Procedure
From gel to publication
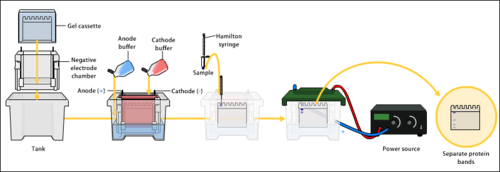
The western blot method is composed of gel electrophoresis to separate native proteins by 3-D structure or denatured proteins by the length of the polypeptide, followed by an electrophoretic transfer onto a membrane (mostly PVDF or nitrocellulose) and an immunostaining procedure to visualize a certain protein on the blot membrane.
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) is generally used for the denaturing electrophoretic separation of proteins. Sodium dodecyl sulfate (SDS) is generally used as a buffer (as well as in the gel) in order to give all proteins present a uniform negative charge, since proteins can be positively, negatively, or neutrally charged. Prior to electrophoresis, protein samples are often boiled to denature the proteins present. This ensures that proteins are separated based on size and prevents proteases (enzymes that break down proteins) from degrading samples. Following electrophoretic separation, the proteins are transferred to a membrane (typically nitrocellulose or PVDF). The membrane is often then stained with Ponceau S in order to visualize the proteins on the blot and ensure a proper transfer occurred. Next the proteins are blocked with milk (or other blocking agents) to prevent non-specific antibody binding, and then stained with antibodies specific to the target protein.[7][6] Lastly, the membrane will be stained with a secondary antibody that recognizes the first antibody staining, which can then be used for detection by a variety of methods. The gel electrophoresis step is included in western blot analysis to resolve the issue of the cross-reactivity of antibodies.
Sample preparation
As a significant step in conducting a western blot, sample preparation has to be done effectively since the interpretation of this assay is influenced by the protein preparation, which is composed of protein extraction and purification processes.[24][3] To achieve efficient protein extraction, a proper homogenization method needs to be chosen due to the fact that it is responsible for bursting the cell membrane and releasing the intracellular components.[3][25] Besides that, the ideal lysis buffer is needed to acquire substantial amounts of target protein content because the buffer is leading the process of protein solubilization and preventing protein degradation. After completing the sample preparation, the protein content is ready to be separated by the utilization of gel electrophoresis.[3]
The proteins of the sample are separated using gel electrophoresis. Separation of proteins may be by isoelectric point (pI), molecular weight, electric charge, or a combination of these factors. The nature of the separation depends on the treatment of the sample and the nature of the gel.
By far the most common type of gel electrophoresis employs polyacrylamide gels and buffers loaded with sodium dodecyl sulfate (SDS). SDS-PAGE (SDS-polyacrylamide gel electrophoresis) maintains polypeptides in a denatured state once they have been treated with strong reducing agents to remove secondary and tertiary structure (e.g. disulfide bonds [S-S] to sulfhydryl groups [SH and SH]) and thus allows separation of proteins by their molecular mass. Sampled proteins become covered in the negatively charged SDS, effectively becoming anionic, and migrate towards the positively charged (higher voltage) anode (usually having a red wire) through the acrylamide mesh of the gel. Smaller proteins migrate faster through this mesh, and the proteins are thus separated according to size (usually measured in kilodaltons, kDa). The concentration of acrylamide determines the resolution of the gel – the greater the acrylamide concentration, the better the resolution of lower molecular weight proteins. The lower the acrylamide concentration, the better the resolution of higher molecular weight proteins. Proteins travel only in one dimension along the gel for most blots.
Samples are loaded into wells in the gel. One lane is usually reserved for a marker or ladder, which is a commercially available mixture of proteins of known molecular weights, typically stained so as to form visible, coloured bands. When voltage is applied along the gel, proteins migrate through it at different speeds dependent on their size. These different rates of advancement (different electrophoretic mobilities) separate into bands within each lane. Protein bands can then be compared to the ladder bands, allowing estimation of the protein's molecular weight.
It is also possible to use a two-dimensional gel which spreads the proteins from a single sample out in two dimensions. Proteins are separated according to isoelectric point (pH at which they have a neutral net charge) in the first dimension, and according to their molecular weight in the second dimension.
Transfer
Western blot transfer
To make the proteins accessible to antibody detection, they are moved from within the gel onto a membrane, a solid support, which is an essential part of the process. There are two types of membrane: nitrocellulose (NC) or polyvinylidene difluoride (PVDF). NC membrane has high affinity for protein and its retention abilities. However, NC is brittle, and does not allow the blot to be used for re-probing, whereas PVDF membrane allows the blot to be re-probed.[1] The most commonly used method for transferring the proteins is called electroblotting. Electroblotting uses an electric current to pull the negatively charged proteins from the gel towards the positively charged anode, and into the PVDF or NC membrane. The proteins move from within the gel onto the membrane while maintaining the organization they had within the gel. An older method of transfer involves placing a membrane on top of the gel, and a stack of filter papers on top of that. The entire stack is placed in a buffer solution which moves up the paper by capillary action, bringing the proteins with it. In practice this method is not commonly used due to the lengthy procedure time.
As a result of either transfer process, the proteins are exposed on a thin membrane layer for detection. Both varieties of membrane are chosen for their non-specific protein binding properties (i.e. binds all proteins equally well). Protein binding is based upon hydrophobic interactions, as well as charged interactions between the membrane and protein. Nitrocellulose membranes are cheaper than PVDF, but are far more fragile and cannot withstand repeated probings.
Total protein staining
Western blot binding
Total protein staining allows the total protein that has been successfully transferred to the membrane to be visualised, allowing the user to check the uniformity of protein transfer and to perform subsequent normalization of the target protein with the actual protein amount per lane. Normalization with the so-called "loading control" was based on immunostaining of housekeeping proteins in the classical procedure, but is heading toward total protein staining recently, due to multiple benefits.[26] At least seven different approaches for total protein staining have been described for western blot normalization: Ponceau S, stain-free techniques, Sypro Ruby, Epicocconone, Coomassie R-350, Amido Black, and Cy5.[26] In order to avoid noise of signal, total protein staining should be performed before blocking of the membrane. Nevertheless, post-antibody stainings have been described as well.[27]
Blocking
Since the membrane has been chosen for its ability to bind protein and as both antibodies and the target are proteins, steps must be taken to prevent the interactions between the membrane and the antibody used for detection of the target protein. Blocking of non-specific binding is achieved by placing the membrane in a dilute solution of protein – typically 3–5% bovine serum albumin (BSA) or non-fat dry milk (both are inexpensive) in tris-buffered saline (TBS) or I-Block, with a minute percentage (0.1%) of detergent such as Tween 20 or Triton X-100. Although non-fat dry milk is preferred due to its availability, an appropriate blocking solution is needed as not all proteins in milk are compatible with all the detection bands.[1] The protein in the dilute solution attaches to the membrane in all places where the target proteins have not attached. Thus, when the antibody is added, it cannot bind to the membrane, and therefore the only available binding site is the specific target protein. This reduces background in the final product of the western blot, leading to clearer results, and eliminates false positives.
Incubation
Probing process
During the detection process, the membrane is "probed" for the protein of interest with a modified antibody which is linked to a reporter enzyme; when exposed to an appropriate substrate, this enzyme drives a colorimetric reaction and produces a colour. For a variety of reasons, this traditionally takes place in a two-step process, although there are now one-step detection methods available for certain applications.
Primary antibody
The primary antibodies are generated when a host species or immune cell culture is exposed to the protein of interest (or a part thereof). Normally, this is part of the immune response, whereas here they are harvested and used as sensitive and specific detection tools that bind the protein directly.
After blocking, a solution of primary antibody (generally between 0.5 and 5 micrograms/mL) diluted in either PBS or TBST wash buffer is incubated with the membrane under gentle agitation for typically an hour at room temperature, or overnight at 4°C. It can also be incubated at different temperatures, with lesser temperatures being associated with more binding, both specific (to the target protein, the "signal") and non-specific ("noise"). Following incubation, the membrane is washed several times in wash buffer to remove unbound primary antibody, and thereby minimize background.[1] Typically, the wash buffer solution is composed of buffered saline solution with a small percentage of detergent, and sometimes with powdered milk or BSA.
Secondary antibody
After rinsing the membrane to remove unbound primary antibody, the membrane is exposed to another antibody known as the secondary antibody. Antibodies come from animal sources (or animal sourced hybridoma cultures). The secondary antibody recognises and binds to the species-specific portion of the primary antibody. Therefore, an anti-mouse secondary antibody will bind to almost any mouse-sourced primary antibody, and can be referred to as an 'anti-species' antibody (e.g. anti-mouse, anti-goat etc.). To allow detection of the target protein, the secondary antibody is commonly linked to biotin or a reporter enzyme such as alkaline phosphatase or horseradish peroxidase. This means that several secondary antibodies will bind to one primary antibody and enhance the signal, allowing the detection of proteins of a much lower concentration than would be visible by SDS-PAGE alone.
Horseradish peroxidase is commonly linked to secondary antibodies to allow the detection of the target protein by chemiluminescence. The chemiluminescent substrate is cleaved by horseradish peroxidase, resulting in the production of luminescence. Therefore, the production of luminescence is proportional to the amount of horseradish peroxidase-conjugated secondary antibody, and therefore, indirectly measures the presence of the target protein. A sensitive sheet of photographic film is placed against the membrane, and exposure to the light from the reaction creates an image of the antibodies bound to the blot. A cheaper but less sensitive approach utilizes a 4-chloronaphthol stain with 1% hydrogen peroxide; the reaction of peroxide radicals with 4-chloronaphthol produces a dark purple stain that can be photographed without using specialized photographic film.
As with the ELISPOT and ELISA procedures, the enzyme can be provided with a substrate molecule that will be converted by the enzyme to a coloured reaction product that will be visible on the membrane (see the figure below with blue bands).
Another method of secondary antibody detection utilizes a near-infrared fluorophore-linked antibody. The light produced from the excitation of a fluorescent dye is static, making fluorescent detection a more precise and accurate measure of the difference in the signal produced by labeled antibodies bound to proteins on a western blot. Proteins can be accurately quantified because the signal generated by the different amounts of proteins on the membranes is measured in a static state, as compared to chemiluminescence, in which light is measured in a dynamic state.[28]
A third alternative is to use a radioactive label rather than an enzyme coupled to the secondary antibody, such as labeling an antibody-binding protein like Staphylococcus Protein A or Streptavidin with a radioactive isotope of iodine. Since other methods are safer, quicker, and cheaper, this method is now rarely used; however, an advantage of this approach is the sensitivity of auto-radiography-based imaging, which enables highly accurate protein quantification when combined with optical software (e.g. Optiquant).
One step
Historically, the probing process was performed in two steps because of the relative ease of producing primary and secondary antibodies in separate processes. This gives researchers and corporations huge advantages in terms of flexibility, reduction of cost, and adds an amplification step to the detection process. Given the advent of high-throughput protein analysis and lower limits of detection, however, there has been interest in developing one-step probing systems that would allow the process to occur faster and with fewer consumables. This requires a probe antibody which both recognizes the protein of interest and contains a detectable label, probes which are often available for known protein tags. The primary probe is incubated with the membrane in a manner similar to that for the primary antibody in a two-step process, and then is ready for direct detection after a series of wash steps.
Detection and visualization
Western blot chemiluminescent detection
After the unbound probes are washed away, the western blot is ready for detection of the probes that are labeled and bound to the protein of interest. In practical terms, not all westerns reveal protein only at one band in a membrane. Size approximations are taken by comparing the stained bands to that of the marker or ladder loaded during electrophoresis. The process is commonly repeated for a structural protein, such as actin or tubulin, that should not change between samples. The amount of target protein is normalized to the structural protein to control between groups. A superior strategy is the normalization to the total protein visualized with trichloroethanol[29][30] or epicocconone.[31] This practice ensures correction for the amount of total protein on the membrane in case of errors or incomplete transfers. (see western blot normalization)
Colorimetric detection
The colorimetric detection method depends on incubation of the western blot with a substrate that reacts with the reporter enzyme (such as peroxidase) that is bound to the secondary antibody. This converts the soluble dye into an insoluble form of a different colour that precipitates next to the enzyme and thereby stains the membrane. Development of the blot is then stopped by washing away the soluble dye. Protein levels are evaluated through densitometry (how intense the stain is) or spectrophotometry.
Chemiluminescent detection
Chemiluminescent detection methods depend on incubation of the western blot with a substrate that will luminesce when exposed to the reporter on the secondary antibody. The light is then detected by CCD cameras which capture a digital image of the western blot or photographic film. The use of film for western blot detection is slowly disappearing because of non linearity of the image (non accurate quantification). The image is analysed by densitometry, which evaluates the relative amount of protein staining and quantifies the results in terms of optical density. Newer software allows further data analysis such as molecular weight analysis if appropriate standards are used.
Radioactive detection
Radioactive labels do not require enzyme substrates, but rather, allow the placement of medical X-ray film directly against the western blot, which develops as it is exposed to the label and creates dark regions which correspond to the protein bands of interest (see image above). The importance of radioactive detections methods is declining due to its hazardous radiation [citation needed], because it is very expensive, health and safety risks are high, and ECL (enhanced chemiluminescence) provides a useful alternative.
Fluorescent detection
Western blot using an anti-lipoic acid primary antibody and an IR-dye labelled secondary antibody in Leishmania major extracts
The fluorescently labeled probe is excited by light and the emission of the excitation is then detected by a photosensor such as a CCD camera equipped with appropriate emission filters which captures a digital image of the western blot and allows further data analysis such as molecular weight analysis and a quantitative western blot analysis. Fluorescence is considered to be one of the best methods for quantification but is less sensitive than chemiluminescence.[32]
Secondary probing
One major difference between nitrocellulose and PVDF membranes relates to the ability of each to support "stripping" antibodies off and reusing the membrane for subsequent antibody probes. While there are well-established protocols available for stripping nitrocellulose membranes, the sturdier PVDF allows for easier stripping, and for more reuse before background noise limits experiments. Another difference is that, unlike nitrocellulose, PVDF must be soaked in 95% ethanol, isopropanol or methanol before use. PVDF membranes also tend to be thicker and more resistant to damage during use.[33]
Minimum requirement specification for Western Blot
In order to ensure that the results of Western blots are reproducible, it is important to report the various parameters mentioned above, including specimen preparation, the concentration of protein used for loading, the percentage of gel and running condition, various transfer methods, attempting to block conditions, the concentration of antibodies, and identification and quantitative determination methods. Many of the articles that have been published don't cover all of these variables. Hence, it is crucial to describe different experimental circumstances or parameters in order to increase the repeatability and precision of WB. To increase WB repeatability, a minimum reporting criteria is thus required.[2][34]
Two-dimensional SDS-PAGE uses the principles and techniques outlined above. 2-D SDS-PAGE, as the name suggests, involves the migration of polypeptides in 2 dimensions. For example, in the first dimension, polypeptides are separated according to isoelectric point, while in the second dimension, polypeptides are separated according to their molecular weight. The isoelectric point of a given protein is determined by the relative number of positively (e.g. lysine, arginine) and negatively (e.g. glutamate, aspartate) charged amino acids, with negatively charged amino acids contributing to a low isoelectric point and positively charged amino acids contributing to a high isoelectric point. Samples could also be separated first under nonreducing conditions using SDS-PAGE, and under reducing conditions in the second dimension, which breaks apart disulfide bonds that hold subunits together. SDS-PAGE might also be coupled with urea-PAGE for a 2-dimensional gel.
In principle, this method allows for the separation of all cellular proteins on a single large gel. A major advantage of this method is that it often distinguishes between different isoforms of a particular protein – e.g. a protein that has been phosphorylated (by addition of a negatively charged group). Proteins that have been separated can be cut out of the gel and then analysed by mass spectrometry, which identifies their molecular weight.
Problems
Detection problems
There may be a weak or absent signal in the band for a number of reasons related to the amount of antibody and antigen used. This problem might be resolved by using the ideal antigen and antibody concentrations and dilutions specified in the supplier's data sheet. Increasing the exposition period in the detection system's software can address weak bands caused by lower sample and antibody concentrations.[2]
Multiple band problems
When the protein is broken down by proteases, several bands other than predicted bands of low molecular weight might appear. The development of numerous bands can be prevented by properly preparing protein samples with enough protease inhibitors. Multiple bands might show up in the high molecular weight region because some proteins form dimers, trimers, and multimers; this issue might be solved by heating the sample for longer periods of time. Proteins with post-translational modifications (PTMs) or numerous isoforms cause several bands to appear at various molecular weight areas. PTMs can be removed from a specimen using specific chemicals, which also remove extra bands.[2]
High background
Strong antibody concentrations, inadequate blocking, inadequate washing, and excessive exposure time during imaging can result in a high background in the blots. A high background in the blots could be avoided by fixing these issues.[2]
Irregular and uneven bands
It has been claimed that a variety of odd and unequal bands, including black dots, white spots or bands, and curving bands, have occurred. The block dots are removed from the blots by effective blocking. White patches develop as a result of bubbles between the membrane and gel. White bands appear in the blots when main and secondary antibodies are present in significant concentrations. Because of the high voltage used during the gel run and the rapid protein migration, smiley bands appear in the blots. The strange bands in the blot are resolved by resolving these problems.[2]
Mitigations
During the western blotting, there could be several problems related to the different steps of this procedure. Those problems could originate from a protein analysis step such as the detection of low- or post-translationally modified proteins. Additionally, they can be based on the selection of antibodies since the quality of the antibodies plays a significant role in the detection of proteins specifically.[3] On account of the presence of these kinds of problems, a variety of improvements are being produced in the fields of preparation of cell lysate and blotting procedures to build up reliable results. Moreover, to achieve more sensitive analysis and overcome the problems associated with western blotting, several different techniques have been developed and utilized, such as far-western blotting, diffusion blotting, single-cell resolution western blotting, and automated microfluidic western blotting.[3]
Presentation
Researchers use different software to process and align image-sections for elegant presentation of western blot results. Popular tools include Sciugo, Microsoft PowerPoint, Adobe Illustrator and GIMP.
↑Burnette WN (April 1981). ""Western blotting": electrophoretic transfer of proteins from sodium dodecyl sulfate--polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A". Analytical Biochemistry. 112 (2): 195–203. doi:10.1016/0003-2697(81)90281-5. PMID6266278.
↑De Castro L, Yoshida CF, Gaspar AM, Gomes SA (December 1996). "Western blot analysis of the reactivity between envelope proteins of hepatitis B viruses from Brazilian carriers and antibodies raised against recombinant hepatitis B vaccines". Acta Virologica. 40 (5–6): 251–258. PMID9171452.
↑Welinder C, Ekblad L (March 2011). "Coomassie staining as loading control in Western blot analysis". Journal of Proteome Research. 10 (3): 1416–1419. doi:10.1021/pr1011476. PMID21186791.
↑Moritz CP, Marz SX, Reiss R, Schulenborg T, Friauf E (February 2014). "Epicocconone staining: a powerful loading control for Western blots". Proteomics. 14 (2–3): 162–168. doi:10.1002/pmic.201300089. PMID24339236. S2CID206368546.
↑Mathews ST, Plaisance EP, Kim T (2009). "Imaging Systems for Westerns: Chemiluminescence vs. Infrared Detection". Protein Blotting and Detection. Methods in Molecular Biology. Vol.536. Humana Press. pp.499–513. doi:10.1007/978-1-59745-542-8_51. ISBN978-1-934115-73-2. PMID19378087.
↑Kurien BT, Scofield RH (2015). "Other Notable Protein Blotting Methods: A Brief Review". In Kurien BT, Scofield RH (eds.). Western Blotting. Methods in Molecular Biology. Vol.1312. New York, NY: Springer New York. pp.487–503. doi:10.1007/978-1-4939-2694-7_51. ISBN978-1-4939-2693-0. PMC7301620. PMID26044032.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.