A signal peptide is a short peptide present at the N-terminus of most newly synthesized proteins that are destined toward the secretory pathway. These proteins include those that reside either inside certain organelles, secreted from the cell, or inserted into most cellular membranes. Although most type I membrane-bound proteins have signal peptides, most type II and multi-spanning membrane-bound proteins are targeted to the secretory pathway by their first transmembrane domain, which biochemically resembles a signal sequence except that it is not cleaved. They are a kind of target peptide.

Tandem mass spectrometry, also known as MS/MS or MS2, is a technique in instrumental analysis where two or more stages of analysis using one or more mass analyzer are performed with an additional reaction step in between these analyses to increase their abilities to analyse chemical samples. A common use of tandem MS is the analysis of biomolecules, such as proteins and peptides.
Edman degradation, developed by Pehr Edman, is a method of sequencing amino acids in a peptide. In this method, the amino-terminal residue is labeled and cleaved from the peptide without disrupting the peptide bonds between other amino acid residues.

Peptide mass fingerprinting (PMF), also known as protein fingerprinting, is an analytical technique for protein identification in which the unknown protein of interest is first cleaved into smaller peptides, whose absolute masses can be accurately measured with a mass spectrometer such as MALDI-TOF or ESI-TOF. The method was developed in 1993 by several groups independently. The peptide masses are compared to either a database containing known protein sequences or even the genome. This is achieved by using computer programs that translate the known genome of the organism into proteins, then theoretically cut the proteins into peptides, and calculate the absolute masses of the peptides from each protein. They then compare the masses of the peptides of the unknown protein to the theoretical peptide masses of each protein encoded in the genome. The results are statistically analyzed to find the best match.

In mass spectrometry, matrix-assisted laser desorption/ionization (MALDI) is an ionization technique that uses a laser energy-absorbing matrix to create ions from large molecules with minimal fragmentation. It has been applied to the analysis of biomolecules and various organic molecules, which tend to be fragile and fragment when ionized by more conventional ionization methods. It is similar in character to electrospray ionization (ESI) in that both techniques are relatively soft ways of obtaining ions of large molecules in the gas phase, though MALDI typically produces far fewer multi-charged ions.
Protein methods are the techniques used to study proteins. There are experimental methods for studying proteins. Computational methods typically use computer programs to analyze proteins. However, many experimental methods require computational analysis of the raw data.

Pehr Victor Edman was a Swedish biochemist. He developed a method for sequencing proteins; the Edman degradation.
A peptide sequence tag is a piece of information about a peptide obtained by tandem mass spectrometry that can be used to identify this peptide in a protein database.
Mascot is a software search engine that uses mass spectrometry data to identify proteins from peptide sequence databases. Mascot is widely used by research facilities around the world. Mascot uses a probabilistic scoring algorithm for protein identification that was adapted from the MOWSE algorithm. Mascot is freely available to use on the website of Matrix Science. A license is required for in-house use where more features can be incorporated.

Electron-transfer dissociation (ETD) is a method of fragmenting multiply-charged gaseous macromolecules in a mass spectrometer between the stages of tandem mass spectrometry (MS/MS). Similar to electron-capture dissociation, ETD induces fragmentation of large, multiply-charged cations by transferring electrons to them. ETD is used extensively with polymers and biological molecules such as proteins and peptides for sequence analysis. Transferring an electron causes peptide backbone cleavage into c- and z-ions while leaving labile post translational modifications (PTM) intact. The technique only works well for higher charge state peptide or polymer ions (z>2). However, relative to collision-induced dissociation (CID), ETD is advantageous for the fragmentation of longer peptides or even entire proteins. This makes the technique important for top-down proteomics. The method was developed by Hunt and coworkers at the University of Virginia.
The Bergmann degradation is a series of chemical reactions designed to remove a single amino acid from the carboxylic acid (C-terminal) end of a peptide. First demonstrated by Max Bergmann in 1934, it is a rarely used method for sequencing peptides. The later developed Edman degradation is an improvement upon the Bergmann degradation, instead cleaving the N-terminal amino acid of peptides to produce a hydantoin containing the desired amino acid.

Protein mass spectrometry refers to the application of mass spectrometry to the study of proteins. Mass spectrometry is an important method for the accurate mass determination and characterization of proteins, and a variety of methods and instrumentations have been developed for its many uses. Its applications include the identification of proteins and their post-translational modifications, the elucidation of protein complexes, their subunits and functional interactions, as well as the global measurement of proteins in proteomics. It can also be used to localize proteins to the various organelles, and determine the interactions between different proteins as well as with membrane lipids.

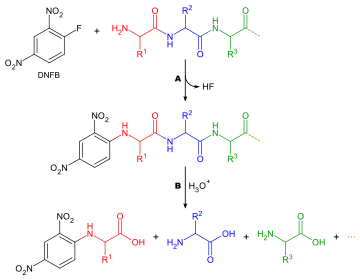
1-Fluoro-2,4-dinitrobenzene is a chemical that reacts with the N-terminal amino acid of polypeptides. This can be helpful for sequencing proteins.
Shotgun proteomics refers to the use of bottom-up proteomics techniques in identifying proteins in complex mixtures using a combination of high performance liquid chromatography combined with mass spectrometry. The name is derived from shotgun sequencing of DNA which is itself named after the rapidly expanding, quasi-random firing pattern of a shotgun. The most common method of shotgun proteomics starts with the proteins in the mixture being digested and the resulting peptides are separated by liquid chromatography. Tandem mass spectrometry is then used to identify the peptides.
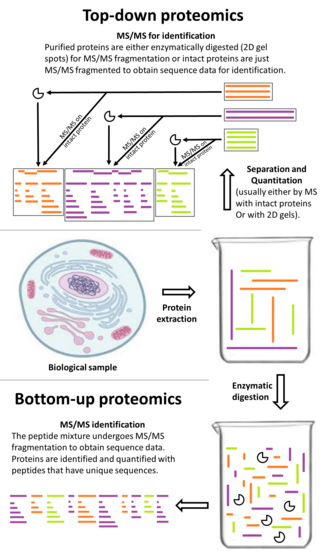
Bottom-up proteomics is a common method to identify proteins and characterize their amino acid sequences and post-translational modifications by proteolytic digestion of proteins prior to analysis by mass spectrometry. The major alternative workflow used in proteomics is called top-down proteomics where intact proteins are purified prior to digestion and/or fragmentation either within the mass spectrometer or by 2D electrophoresis. Essentially, bottom-up proteomics is a relatively simple and reliable means of determining the protein make-up of a given sample of cells, tissues, etc.

Isobaric tags for relative and absolute quantitation (iTRAQ) is an isobaric labeling method used in quantitative proteomics by tandem mass spectrometry to determine the amount of proteins from different sources in a single experiment. It uses stable isotope labeled molecules that can be covalent bonded to the N-terminus and side chain amines of proteins.
An isotope-coded affinity tag (ICAT) is an in-vitro isotopic labeling method used for quantitative proteomics by mass spectrometry that uses chemical labeling reagents. These chemical probes consist of three elements: a reactive group for labeling an amino acid side chain, an isotopically coded linker, and a tag for the affinity isolation of labeled proteins/peptides. The samples are combined and then separated through chromatography, then sent through a mass spectrometer to determine the mass-to-charge ratio between the proteins. Only cysteine containing peptides can be analysed. Since only cysteine containing peptides are analysed, often the post translational modification is lost.
Terminal amine isotopic labeling of substrates (TAILS) is a method in quantitative proteomics that identifies the protein content of samples based on N-terminal fragments of each protein and detects differences in protein abundance among samples.
In mass spectrometry, de novo peptide sequencing is the method in which a peptide amino acid sequence is determined from tandem mass spectrometry.

Ancient proteins are complex mixtures and the term palaeoproteomics is used to characterise the study of proteomes in the past. Ancients proteins have been recovered from a wide range of archaeological materials, including bones, teeth, eggshells, leathers, parchments, ceramics, painting binders and well-preserved soft tissues like gut intestines. These preserved proteins have provided valuable information about taxonomic identification, evolution history (phylogeny), diet, health, disease, technology and social dynamics in the past.