Knowing the amino acid sequence of peptides from a protein digest is essential for studying the biological function of the protein. In the old days, this was accomplished by the Edman degradation procedure.[1] Today, analysis by a tandem mass spectrometer is a more common method to solve the sequencing of peptides. Generally, there are two approaches: database search and de novo sequencing. Database search is a simple version as the mass spectra data of the unknown peptide is submitted and run to find a match with a known peptide sequence, the peptide with the highest matching score will be selected.[2] This approach fails to recognize novel peptides since it can only match to existing sequences in the database. De novo sequencing is an assignment of fragment ions from a mass spectrum. Different algorithms[3] are used for interpretation and most instruments come with de novo sequencing programs.
Peptide fragmentation
Peptides are protonated in positive-ion mode. The proton initially locates at the N-terminus or a basic residue side chain, but because of the internal solvation, it can move along the backbone breaking at different sites which result in different fragments. The fragmentation rules are well explained by some publications.[4][5][6][7][8][9]
Three different types of backbone bonds can be broken to form peptide fragments: alkyl carbonyl (CHR-CO), peptide amide bond (CO-NH), and amino alkyl bond (NH-CHR).[citation needed]
Different types of fragment ions
6 types of sequence ions in peptide fragmentation
When the backbone bonds cleave, six different types of sequence ions are formed as shown in Fig. 1. The N-terminal charged fragment ions are classed as a, b or c, while the C-terminal charged ones are classed as x, y or z. The subscript n is the number of amino acid residues. The nomenclature was first proposed by Roepstorff and Fohlman, then Biemann modified it and this became the most widely accepted version.[11][12]
Among these sequence ions, a, b and y-ions are the most common ion types, especially in the low-energy collision-induced dissociation (CID) mass spectrometers, since the peptide amide bond (CO-NH) is the most vulnerable and the loss of CO from b-ions.
Mass of b-ions = Σ (residue masses) + 1 (H+)
Mass of y-ions = Σ (residue masses) + 19 (H2O+H+)
Mass of a-ions = mass of b-ions – 28 (CO)
Double backbone cleavage produces internal ions, acylium-type like H2N-CHR2-CO-NH-CHR3-CO+ or immonium-type like H2N-CHR2-CO-NH+=CHR3. These ions are usually disturbance in the spectra.
Satellite ions in peptide fragmentation
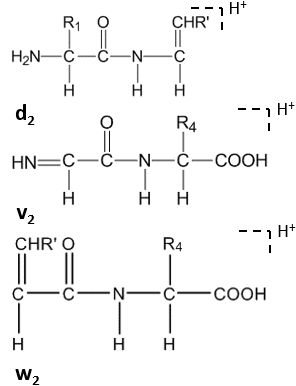
Further cleavage happens under high-energy CID at the side chain of C-terminal residues, forming dn, vn, wn-ions.[8]
Fragmentation rules summary
Most fragment ions are b- or y-ions. a-ions are also frequently seen by the loss of CO from b-ions.[9]
Satellite ions(wn, vn, dn-ions) are formed by high-energy CID.
Ser-, Thr-, Asp- and Glu-containing ions generate neutral molecular loss of water (-18).
Asn-, Gln-, Lys-, Arg-containing ions generate neutral molecular loss of ammonia (-17).
Neutral loss of ammonia from Arg leads to fragment ions (y-17) or (b-17) ions with higher abundance than their corresponding ions.
When C-terminus has a basic residue, the peptide generates (bn-1+18) ion.
A complementary b-y ion pair can be observed in multiply charged ions spectra. For this b-y ion pair, the sum of their subscripts is equal to the total number of amino acid residues in the unknown peptide.
If the C-terminus is Arg or Lys, y1-ion can be found in the spectrum to prove it.
Methods for peptide fragmentation
In low energy collision induced dissociation (CID), b- and y-ions are the main product ions. In addition, loss of ammonia (-17 Da) is observed in fragment with RKNQ amino acids in it. Loss of water (-18 Da) can be observed in fragment with STED amino acids in it. No satellite ions are shown in the spectra.[citation needed]
In high energy CID, all different types of fragment ions can be observed but no losses of ammonia or water.[citation needed]
For post source decay (PSD) in MALDI, a, b, y-ions are most common product ions.[citation needed]
Factors affecting fragmentation are the charge state (the higher charge state, the less energy is needed for fragmentation), mass of the peptide (the larger mass, the more energy is required), induced energy (higher energy leads to more fragmentation), primary amino acid sequence, mode of dissociation and collision gas.[citation needed]
Guidelines for interpretation
Table 1. Mass of amino acid fragment ions
For interpretation,[14] first, look for single amino acid immonium ions (H2N+=CHR2). Corresponding immonium ions for amino acids are listed in Table 1. Ignore a few peaks at the high-mass end of the spectrum. They are ions that undergo neutral molecules losses (H2O, NH3, CO2, HCOOH) from [M+H]+ ions. Find mass differences at 28 Da since b-ions can form a-ions by loss of CO. Look for b2-ions at low-mass end of the spectrum, which helps to identify yn-2-ions too. Mass of b2-ions are listed in Table 2, as well as single amino acids that have equal mass to b2-ions.[15] The mass of b2-ion = mass of two amino acid residues + 1.
Table 2. Mass of b2-ions in peptide fragmentation
Identify a sequence ion series by the same mass difference, which matches one of the amino acid residue masses (see Table 1). For example, mass differences between an and an-1, bn and bn-1, cn and cn-1 are the same. Identify yn-1-ion at the high-mass end of the spectrum. Then continue to identify yn-2, yn-3... ions by matching mass differences with the amino acid residue masses (see Table 1). Look for the corresponding b-ions of the identified y-ions. The mass of b+y ions is the mass of the peptide +2 Da. After identifying the y-ion series and b-ion series, assign the amino acid sequence and check the mass. The other method is to identify b-ions first and then find the corresponding y-ions.[citation needed]
Algorithms and software
Manual de novo sequencing is labor-intensive and time-consuming. Usually algorithms or programs come with the mass spectrometer instrument are applied for the interpretation of spectra.
Development of de novo sequencing algorithms
An old method is to list all possible peptides for the precursor ion in mass spectrum, and match the mass spectrum for each candidate to the experimental spectrum. The possible peptide that has the most similar spectrum will have the highest chance to be the right sequence. However, the number of possible peptides may be large. For example, a precursor peptide with a molecular weight of 774 has 21,909,046 possible peptides. Even though it is done in the computer, it takes a long time.[17][18]
Another method is called "subsequencing", which instead of listing whole sequence of possible peptides, matches short sequences of peptides that represent only a part of the complete peptide. When sequences that highly match the fragment ions in the experimental spectrum are found, they are extended by residues one by one to find the best matching.[19][20][21][22]
In the third method, graphical display of the data is applied, in which fragment ions that have the same mass differences of one amino acid residue are connected by lines. In this way, it is easier to get a clear image of ion series of the same type. This method could be helpful for manual de novo peptide sequencing, but doesn't work for high-throughput condition.[23]
The fourth method, which is considered to be successful, is the graph theory. Applying graph theory in de novo peptide sequencing was first mentioned by Bartels.[24] Peaks in the spectrum are transformed into vertices in a graph called "spectrum graph". If two vertices have the same mass difference of one or several amino acids, a directed edge will be applied. The SeqMS algorithm,[25] Lutefisk algorithm,[26] Sherenga algorithm[27] are some examples of this type.
Deep Learning
More recently, deep learning techniques have been applied to solve the de novo peptide sequencing problem. The first breakthrough was DeepNovo, which adopted the convolutional neural network structure, achieved major improvements in sequence accuracy, and enabled complete protein sequence assembly without assisting databases[28] Subsequently, additional network structures, such as PointNet (PointNovo[29]), have been adopted to extract features from a raw spectrum. The de novo peptide sequencing problem is then framed as a sequence prediction problem. Given previously predicted partial peptide sequence, neural-network-based de novo peptide sequencing models will repeatedly generate the most probable next amino acid until the predicted peptide's mass matches the precursor mass. At inference time, search strategies such as beam search can be adopted to explore a larger search space while keeping the computational cost low. Comparing with previous methods, neural-network-based models have demonstrated significantly better accuracy and sensitivity.[28][29][30] Moreover, with a careful model design, deep-learning-based de novo peptide sequencing algorithms can also be fast enough to achieve real-time peptide de novo sequencing.[29] PEAKS software incorporates this neural network learning in their de novo sequencing algorithms.
Software packages
As described by Andreotti et al. in 2012,[31] Antilope is a combination of Lagrangian relaxation and an adaptation of Yen's k shortest paths. It is based on 'spectrum graph' method and contains different scoring functions, and can be comparable on the running time and accuracy to "the popular state-of-the-art programs" PepNovo and NovoHMM.
Grossmann et al.[32] presented AUDENS in 2005 as an automated de novo peptide sequencing tool containing a preprocessing module that can recognize signal peaks and noise peaks.
Lutefisk can solve de novo sequencing from CID mass spectra. In this algorithm, significant ions are first found, then determine the N- and C-terminal evidence list. Based on the sequence list, it generates complete sequences in spectra and scores them with the experimental spectrum. However, the result may include several sequence candidates that have only little difference, so it is hard to find the right peptide sequence. A second program, CIDentify, which is a modified version by Alex Taylor of Bill Pearson's FASTA algorithm, can be applied to distinguish those uncertain similar candidates.[citation needed]
Mo et al. presented the MSNovo algorithm in 2007 and proved that it performed "better than existing de novo tools on multiple data sets".[33] This algorithm can do de novo sequencing interpretation of LCQ, LTQ mass spectrometers and of singly, doubly, triply charged ions. Different from other algorithms, it applied a novel scoring function and use a mass array instead of a spectrum graph.
Fisher et al.[34] proposed the NovoHMM method of de novo sequencing. A hidden Markov model (HMM) is applied as a new way to solve de novo sequencing in a Bayesian framework. Instead of scoring for single symbols of the sequence, this method considers posterior probabilities for amino acids. In the paper, this method is proved to have better performance than other popular de novo peptide sequencing methods like PepNovo by a lot of example spectra.
PEAKS is a complete software package for the interpretation of peptide mass spectra. It contains de novo sequencing, database search, PTM identification, homology search and quantification in data analysis. Ma et al. described a new model and algorithm for de novo sequencing in PEAKS, and compared the performance with Lutefisk of several tryptic peptides of standard proteins, by the quadrupoletime-of-flight (Q-TOF) mass spectrometer.[35]
PepNovo is a high throughput de novo peptide sequencing tool and uses a probabilistic network as scoring method. It usually takes less than 0.2 seconds for interpretation of one spectrum. Described by Frank et al., PepNovo works better than several popular algorithms like Sherenga, PEAKS, Lutefisk.[36] Now a new version PepNovo+ is available.
Chi et al. presented pNovo+ in 2013 as a new de novo peptide sequencing tool by using complementary HCD and ETD tandem mass spectra.[37] In this method, a component algorithm, pDAG, largely speeds up the acquisition time of peptide sequencing to 0.018s on average, which is three times as fast as the other popular de novo sequencing software.
As described by Jeong et al., compared with other do novo peptide sequencing tools, which works well on only certain types of spectra, UniNovo is a more universal tool that has a good performance on various types of spectra or spectral pairs like CID, ETD, HCD, CID/ETD, etc. It has a better accuracy than PepNovo+ or PEAKS. Moreover, it generates the error rate of the reported peptide sequences.[38]
Ma published Novor in 2015 as a real-time de novo peptide sequencing engine. The tool is sought to improve the de novo speed by an order of magnitude and retain similar accuracy as other de novo tools in the market. On a Macbook Pro laptop, Novor has achieved more than 300 MS/MS spectra per second.[39]
Pevtsov et al. compared the performance of the above five de novo sequencing algorithms: AUDENS, Lutefisk, NovoHMM, PepNovo, and PEAKS . QSTAR and LCQ mass spectrometer data were employed in the analysis, and evaluated by relative sequence distance (RSD) value, which was the similarity between de novo peptide sequencing and true peptide sequence calculated by a dynamic programming method. Results showed that all algorithms had better performance in QSTAR data than on LCQ data, while PEAKS as the best had a success rate of 49.7% in QSTAR data, and NovoHMM as the best had a success rate of 18.3% in LCQ data. The performance order in QSTAR data was PEAKS > Lutefisk, PepNovo > AUDENS, NovoHMM, and in LCQ data was NovoHMM > PepNovo, PEAKS > Lutefisk > AUDENS. Compared in a range of spectrum quality, PEAKS and NovoHMM also showed the best performance in both data among all 5 algorithms. PEAKS and NovoHMM had the best sensitivity in both QSTAR and LCQ data as well. However, no evaluated algorithms exceeded a 50% of exact identification for both data sets.[40]
Recent progress in mass spectrometers made it possible to generate mass spectra of ultra-high resolution . The improved accuracy, together with the increased amount of mass spectrometry data that are being generated, draws the interests of applying deep learning techniques to de novo peptide sequencing. In 2017 Tran et al. proposed DeepNovo, the first deep learning based de novo sequencing software. The benchmark analysis in the original publication demonstrated that DeepNovo outperformed previous methods, including PEAKS, Novor and PepNovo, by a significant margin. DeepNovo is implemented in python with the Tensorflow framework.[41] To represent a mass spectrum as a fixed-dimensional input to the neural-network, DeepNovo discretized each spectrum into a length 150,000 vector. This unnecessarily large spectrum representation, and the single-thread CPU usage in the original implementation, prevents DeepNovo from performing peptide sequencing in real time. To further improve efficiency of de novo peptide sequencing models, Qiao et al. proposed PointNovo in 2020. PointNovo is a python software implemented with the PyTorch framework [42] and it gets rid of the space consuming spectrum-vector-representation adopted by DeepNovo. Comparing with DeepNovo, PointNovo managed to achieve better accuracy and efficiency at the same time by directly representing a spectrum as a set of m/z and intensity pairs.[citation needed]
Related Research Articles
Mass spectrometry (MS) is an analytical technique that is used to measure the mass-to-charge ratio of ions. The results are presented as a mass spectrum, a plot of intensity as a function of the mass-to-charge ratio. Mass spectrometry is used in many different fields and is applied to pure samples as well as complex mixtures.
Tandem mass spectrometry, also known as MS/MS or MS2, is a technique in instrumental analysis where two or more stages of analysis using one or more mass analyzer are performed with an additional reaction step in between these analyses to increase their abilities to analyse chemical samples. A common use of tandem MS is the analysis of biomolecules, such as proteins and peptides.
Protein sequencing is the practical process of determining the amino acid sequence of all or part of a protein or peptide. This may serve to identify the protein or characterize its post-translational modifications. Typically, partial sequencing of a protein provides sufficient information to identify it with reference to databases of protein sequences derived from the conceptual translation of genes.
Peptide mass fingerprinting (PMF), also known as protein fingerprinting, is an analytical technique for protein identification in which the unknown protein of interest is first cleaved into smaller peptides, whose absolute masses can be accurately measured with a mass spectrometer such as MALDI-TOF or ESI-TOF. The method was developed in 1993 by several groups independently. The peptide masses are compared to either a database containing known protein sequences or even the genome. This is achieved by using computer programs that translate the known genome of the organism into proteins, then theoretically cut the proteins into peptides, and calculate the absolute masses of the peptides from each protein. They then compare the masses of the peptides of the unknown protein to the theoretical peptide masses of each protein encoded in the genome. The results are statistically analyzed to find the best match.
Fast atom bombardment (FAB) is an ionization technique used in mass spectrometry in which a beam of high energy atoms strikes a surface to create ions. It was developed by Michael Barber at the University of Manchester in 1980. When a beam of high energy ions is used instead of atoms, the method is known as liquid secondary ion mass spectrometry (LSIMS). In FAB and LSIMS, the material to be analyzed is mixed with a non-volatile chemical protection environment, called a matrix, and is bombarded under vacuum with a high energy atomic beam. The atoms are typically from an inert gas such as argon or xenon. Common matrices include glycerol, thioglycerol, 3-nitrobenzyl alcohol (3-NBA), 18-crown-6 ether, 2-nitrophenyloctyl ether, sulfolane, diethanolamine, and triethanolamine. This technique is similar to secondary ion mass spectrometry and plasma desorption mass spectrometry.
Sequest is a tandem mass spectrometry data analysis program used for protein identification. Sequest identifies collections of tandem mass spectra to peptide sequences that have been generated from databases of protein sequences.
Hydrogen–deuterium exchange is a chemical reaction in which a covalently bonded hydrogen atom is replaced by a deuterium atom, or vice versa. It can be applied most easily to exchangeable protons and deuterons, where such a transformation occurs in the presence of a suitable deuterium source, without any catalyst. The use of acid, base or metal catalysts, coupled with conditions of increased temperature and pressure, can facilitate the exchange of non-exchangeable hydrogen atoms, so long as the substrate is robust to the conditions and reagents employed. This often results in perdeuteration: hydrogen-deuterium exchange of all non-exchangeable hydrogen atoms in a molecule.
A peptide sequence tag is a piece of information about a peptide obtained by tandem mass spectrometry that can be used to identify this peptide in a protein database.
Mascot is a software search engine that uses mass spectrometry data to identify proteins from peptide sequence databases. Mascot is widely used by research facilities around the world. Mascot uses a probabilistic scoring algorithm for protein identification that was adapted from the MOWSE algorithm. Mascot is freely available to use on the website of Matrix Science. A license is required for in-house use where more features can be incorporated.
PEAKS is a proteomics software program for tandem mass spectrometry designed for peptide sequencing, protein identification and quantification.
Electron-transfer dissociation (ETD) is a method of fragmenting multiply-charged gaseous macromolecules in a mass spectrometer between the stages of tandem mass spectrometry (MS/MS). Similar to electron-capture dissociation, ETD induces fragmentation of large, multiply-charged cations by transferring electrons to them. ETD is used extensively with polymers and biological molecules such as proteins and peptides for sequence analysis. Transferring an electron causes peptide backbone cleavage into c- and z-ions while leaving labile post translational modifications (PTM) intact. The technique only works well for higher charge state peptide or polymer ions (z>2). However, relative to collision-induced dissociation (CID), ETD is advantageous for the fragmentation of longer peptides or even entire proteins. This makes the technique important for top-down proteomics. The method was developed by Hunt and coworkers at the University of Virginia.
Protein mass spectrometry refers to the application of mass spectrometry to the study of proteins. Mass spectrometry is an important method for the accurate mass determination and characterization of proteins, and a variety of methods and instrumentations have been developed for its many uses. Its applications include the identification of proteins and their post-translational modifications, the elucidation of protein complexes, their subunits and functional interactions, as well as the global measurement of proteins in proteomics. It can also be used to localize proteins to the various organelles, and determine the interactions between different proteins as well as with membrane lipids.
Shotgun proteomics refers to the use of bottom-up proteomics techniques in identifying proteins in complex mixtures using a combination of high performance liquid chromatography combined with mass spectrometry. The name is derived from shotgun sequencing of DNA which is itself named after the rapidly expanding, quasi-random firing pattern of a shotgun. The most common method of shotgun proteomics starts with the proteins in the mixture being digested and the resulting peptides are separated by liquid chromatography. Tandem mass spectrometry is then used to identify the peptides.
Quantitative proteomics is an analytical chemistry technique for determining the amount of proteins in a sample. The methods for protein identification are identical to those used in general proteomics, but include quantification as an additional dimension. Rather than just providing lists of proteins identified in a certain sample, quantitative proteomics yields information about the physiological differences between two biological samples. For example, this approach can be used to compare samples from healthy and diseased patients. Quantitative proteomics is mainly performed by two-dimensional gel electrophoresis (2-DE), preparative native PAGE, or mass spectrometry (MS). However, a recent developed method of quantitative dot blot (QDB) analysis is able to measure both the absolute and relative quantity of an individual proteins in the sample in high throughput format, thus open a new direction for proteomic research. In contrast to 2-DE, which requires MS for the downstream protein identification, MS technology can identify and quantify the changes.
John R. Yates III is an American chemist and Ernest W. Hahn Professor in the Departments of Molecular Medicine and Neurobiology at The Scripps Research Institute in La Jolla, California.
Lys-N is a metalloendopeptidase found in the mushroom Grifola frondosa that cleaves proteins on the amino side of lysine residues.
In bio-informatics, a peptide-mass fingerprint or peptide-mass map is a mass spectrum of a mixture of peptides that comes from a digested protein being analyzed. The mass spectrum serves as a fingerprint in the sense that it is a pattern that can serve to identify the protein. The method for forming a peptide-mass fingerprint, developed in 1993, consists of isolating a protein, breaking it down into individual peptides, and determining the masses of the peptides through some form of mass spectrometry. Once formed, a peptide-mass fingerprint can be used to search in databases for related protein or even genomic sequences, making it a powerful tool for annotation of protein-coding genes.
In mass spectrometry, data-independent acquisition (DIA) is a method of molecular structure determination in which all ions within a selected m/z range are fragmented and analyzed in a second stage of tandem mass spectrometry. Tandem mass spectra are acquired either by fragmenting all ions that enter the mass spectrometer at a given time or by sequentially isolating and fragmenting ranges of m/z. DIA is an alternative to data-dependent acquisition (DDA) where a fixed number of precursor ions are selected and analyzed by tandem mass spectrometry.
Ancient proteins are complex mixtures and the term palaeoproteomics is used to characterise the study of proteomes in the past. Ancients proteins have been recovered from a wide range of archaeological materials, including bones, teeth, eggshells, leathers, parchments, ceramics, painting binders and well-preserved soft tissues like gut intestines. These preserved proteins have provided valuable information about taxonomic identification, evolution history (phylogeny), diet, health, disease, technology and social dynamics in the past.
↑ Lu, Bingwen; Chen, Ting (March 2004). "Algorithms for de novo peptide sequencing using tandem mass spectrometry". Drug Discovery Today: BIOSILICO. 2 (2): 85–90. doi:10.1016/S1741-8364(04)02387-X.
1 2 Papayannopoulos, Ioannis A. (January 1995). "The interpretation of collision-induced dissociation tandem mass spectra of peptides". Mass Spectrometry Reviews. 14 (1): 49–73. Bibcode:1995MSRv...14...49P. doi:10.1002/mas.1280140104.
↑ Dass, Chhabil; Desiderio, Dominic M. (May 1987). "Fast atom bombardment mass spectrometry analysis of opioid peptides". Analytical Biochemistry. 163 (1): 52–66. doi:10.1016/0003-2697(87)90092-3. PMID2887130.
↑ Tang, Xue-Jun; Boyd, Robert K.; Bertrand, M. J. (November 1992). "An investigation of fragmentation mechanisms of doubly protonated tryptic peptides". Rapid Communications in Mass Spectrometry. 6 (11): 651–657. Bibcode:1992RCMS....6..651T. doi:10.1002/rcm.1290061105. PMID1467549.
1 2 3 Johnson, Richard S.; Martin, Stephen A.; Biemann, Klaus (December 1988). "Collision-induced fragmentation of (M + H)+ ions of peptides. Side chain specific sequence ions". International Journal of Mass Spectrometry and Ion Processes. 86: 137–154. Bibcode:1988IJMSI..86..137J. doi:10.1016/0168-1176(88)80060-0.
1 2 Dass, Chhabil (2007). Fundamentals of contemporary mass spectrometry ([Online-Ausg.].ed.). Hoboken, N.J.: Wiley-Interscience. pp.317–322. doi:10.1002/0470118490. ISBN9780470118498.
↑ Dass, Chhabil (2001). Principles and practice of biological mass spectrometry. New York, NY [u.a.]: Wiley. ISBN978-0-471-33053-0.
↑ Roepstorff, P; Fohlman, J (November 1984). "Proposal for a common nomenclature for sequence ions in mass spectra of peptides". Biomedical Mass Spectrometry. 11 (11): 601. doi:10.1002/bms.1200111109. PMID6525415.
↑ McCloskey, James A., ed. (1990). Mass spectrometry. San Diego: Academic Press. pp.886–887. ISBN978-0121820947.
↑ Dass, Chhabil (2007). Fundamentals of contemporary mass spectrometry ([Online-Ausg.].ed.). Hoboken, N.J.: Wiley-Interscience. pp.327–330. ISBN9780470118498.
↑ Harrison, Alex G.; Csizmadia, Imre G.; Tang, Ting-Hua (May 2000). "Structure and fragmentation of b2 ions in peptide mass spectra". Journal of the American Society for Mass Spectrometry. 11 (5): 427–436. doi:10.1016/S1044-0305(00)00104-5. PMID10790847. S2CID24794690.
↑ Dass, Chhabil (2007). Fundamentals of contemporary mass spectrometry ([Online-Ausg.].ed.). Hoboken, N.J.: Wiley-Interscience. p.329. ISBN9780470118498.
↑ Sakurai, T.; Matsuo, T.; Matsuda, H.; Katakuse, I. (August 1984). "PAAS 3: A computer program to determine probable sequence of peptides from mass spectrometric data". Biological Mass Spectrometry. 11 (8): 396–399. doi:10.1002/bms.1200110806.
↑ Biemann, K; Cone, C; Webster, BR; Arsenault, GP (5 December 1966). "Determination of the amino acid sequence in oligopeptides by computer interpretation of their high-resolution mass spectra". Journal of the American Chemical Society. 88 (23): 5598–606. doi:10.1021/ja00975a045. PMID5980176.
↑ Ishikawa, K.; Niwa, Y. (July 1986). "Computer-aided peptide sequencing by fast atom bombardment mass spectrometry". Biological Mass Spectrometry. 13 (7): 373–380. doi:10.1002/bms.1200130709.
↑ Siegel, MM; Bauman, N (15 March 1988). "An efficient algorithm for sequencing peptides using fast atom bombardment mass spectral data". Biomedical & Environmental Mass Spectrometry. 15 (6): 333–43. doi:10.1002/bms.1200150606. PMID2967723.
↑ Johnson, RS; Biemann, K (November 1989). "Computer program (SEQPEP) to aid in the interpretation of high-energy collision tandem mass spectra of peptides". Biomedical & Environmental Mass Spectrometry. 18 (11): 945–57. doi:10.1002/bms.1200181102. PMID2620156.
↑ Scoble, Hubert A.; Biller, James E.; Biemann, Klaus (1987). "A graphics display-oriented strategy for the amino acid sequencing of peptides by tandem mass spectrometry". Fresenius' Zeitschrift für Analytische Chemie. 327 (2): 239–245. doi:10.1007/BF00469824. S2CID97665981.
↑ Bartels, Christian (June 1990). "Fast algorithm for peptide sequencing by mass spectroscopy". Biological Mass Spectrometry. 19 (6): 363–368. doi:10.1002/bms.1200190607. PMID24730078.
↑ Fernández-de-Cossío, J; Gonzalez, J; Besada, V (August 1995). "A computer program to aid the sequencing of peptides in collision-activated decomposition experiments". Computer Applications in the Biosciences. 11 (4): 427–34. doi:10.1093/bioinformatics/11.4.427. PMID8521052.
↑ Dančík, Vlado; Addona, Theresa A.; Clauser, Karl R.; Vath, James E.; Pevzner, Pavel A. (October 1999). "Peptide Sequencing via Tandem Mass Spectrometry". Journal of Computational Biology. 6 (3–4): 327–342. CiteSeerX10.1.1.128.2645. doi:10.1089/106652799318300. PMID10582570.
1 2 Tran, Ngoc Hieu, etal. "De novo peptide sequencing by deep learning." Proceedings of the National Academy of Sciences 114.31 (2017): 8247-8252.
1 2 3 Qiao, Rui, et al. "Computationally instrument-resolution-independent de novo peptide sequencing for high-resolution devices." Nature Machine Intelligence 3.5 (2021): 420-425.
↑ Karunratanakul, Korrawe, et al. "Uncovering thousands of new peptides with sequence-mask-search hybrid de novo peptide sequencing framework." Molecular & Cellular Proteomics 18.12 (2019): 2478-2491.
↑ Andreotti, S; Klau, GW; Reinert, K (2012). "Antilope--a Lagrangian relaxation approach to the de novo peptide sequencing problem". IEEE/ACM Transactions on Computational Biology and Bioinformatics. 9 (2): 385–94. arXiv:1102.4016. doi:10.1109/tcbb.2011.59. PMID21464512. S2CID593303.
↑ Grossmann, J; Roos, FF; Cieliebak, M; Lipták, Z; Mathis, LK; Müller, M; Gruissem, W; Baginsky, S (2005). "AUDENS: a tool for automated peptide de novo sequencing". Journal of Proteome Research. 4 (5): 1768–74. CiteSeerX10.1.1.654.169. doi:10.1021/pr050070a. PMID16212431.
↑ Mo, L; Dutta, D; Wan, Y; Chen, T (1 July 2007). "MSNovo: a dynamic programming algorithm for de novo peptide sequencing via tandem mass spectrometry". Analytical Chemistry. 79 (13): 4870–8. doi:10.1021/ac070039n. PMID17550227.
↑ Fischer, B; Roth, V; Roos, F; Grossmann, J; Baginsky, S; Widmayer, P; Gruissem, W; Buhmann, JM (15 November 2005). "NovoHMM: a hidden Markov model for de novo peptide sequencing". Analytical Chemistry. 77 (22): 7265–73. CiteSeerX10.1.1.507.1610. doi:10.1021/ac0508853. PMID16285674.
↑ Ma, Bin; Zhang, Kaizhong; Hendrie, Christopher; Liang, Chengzhi; Li, Ming; Doherty-Kirby, Amanda; Lajoie, Gilles (30 October 2003). "PEAKS: powerful software for peptidede novo sequencing by tandem mass spectrometry". Rapid Communications in Mass Spectrometry. 17 (20): 2337–2342. Bibcode:2003RCMS...17.2337M. doi:10.1002/rcm.1196. PMID14558135.
↑ Frank, A; Pevzner, P (15 February 2005). "PepNovo: de novo peptide sequencing via probabilistic network modeling". Analytical Chemistry. 77 (4): 964–73. doi:10.1021/ac048788h. PMID15858974.
↑ Chi, H; Chen, H; He, K; Wu, L; Yang, B; Sun, RX; Liu, J; Zeng, WF; Song, CQ; He, SM; Dong, MQ (1 February 2013). "pNovo+: de novo peptide sequencing using complementary HCD and ETD tandem mass spectra". Journal of Proteome Research. 12 (2): 615–25. doi:10.1021/pr3006843. PMID23272783.
↑ Pevtsov, S.; Fedulova, I.; Mirzaei, H.; Buck, C.; Zhang, X. (2006). "Performance Evaluation of Existing De Novo Sequencing Algorithms". Journal of Proteome Research. 5 (11): 3018–3028. doi:10.1021/pr060222h. PMID17081053.
↑ Abadi, Martín, et al. "Tensorflow: A system for large-scale machine learning." 12th {USENIX} symposium on operating systems design and implementation ({OSDI} 16). 2016.
↑ Adam, et al. "Pytorch: An imperative style, high-performance deep learning library." Advances in neural information processing systems 32 (2019): 8026-8037.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.