Newborn screening (NBS) is a public health program of screening in infants shortly after birth for conditions that are treatable, but not clinically evident in the newborn period. The goal is to identify infants at risk for these conditions early enough to confirm the diagnosis and provide intervention that will alter the clinical course of the disease and prevent or ameliorate the clinical manifestations. NBS started with the discovery that the amino acid disorder phenylketonuria (PKU) could be treated by dietary adjustment, and that early intervention was required for the best outcome. Infants with PKU appear normal at birth, but are unable to metabolize the essential amino acid phenylalanine, resulting in irreversible intellectual disability. In the 1960s, Robert Guthrie developed a simple method using a bacterial inhibition assay that could detect high levels of phenylalanine in blood shortly after a baby was born. Guthrie also pioneered the collection of blood on filter paper which could be easily transported, recognizing the need for a simple system if the screening was going to be done on a large scale. Newborn screening around the world is still done using similar filter paper. NBS was first introduced as a public health program in the United States in the early 1960s, and has expanded to countries around the world.
Screening programs are often run by state or national governing bodies with the goal of screening all infants born in the jurisdiction for a defined panel of treatable disorders. The number of diseases screened for is set by each jurisdiction, and can vary greatly. Most NBS tests are done by measuring metabolites or enzyme activity in whole blood samples collected on filter paper. Bedside tests for hearing loss using automated auditory brainstem response and congenital heart defects using pulse oximetry are included in some NBS programs. Infants who screen positive undergo further testing to determine if they are truly affected with a disease or if the test result was a false positive. Follow-up testing is typically coordinated between geneticists and the infant's pediatrician or primary care physician.
History
Robert Guthrie is given much of the credit for pioneering the earliest screening for phenylketonuria in the late 1960s using a bacterial inhibition assay (BIA) to measure phenylalanine levels in blood samples obtained by pricking a newborn baby's heel on the second day of life on filter paper.[1]Congenital hypothyroidism was the second disease widely added in the 1970s.[2] Guthrie and colleagues also developed bacterial inhibition assays for the detection of maple syrup urine disease and classic galactosemia.[3] The development of tandem mass spectrometry (MS/MS) screening in the early 1990s led to a large expansion of potentially detectable congenital metabolic diseases that can be identified by characteristic patterns of amino acids and acylcarnitines.[4] In many regions, Guthrie's BIA has been replaced by MS/MS profiles, however the filter paper he developed is still used worldwide, and has allowed for the screening of millions of infants around the world each year.[5]
In the United States, the American College of Medical Genetics recommended a uniform panel of diseases that all infants born in every state should be screened for. They also developed an evidence-based review process for the addition of conditions in the future. The implementation of this panel across the United States meant all babies born would be screened for the same number of conditions. This recommendation is not binding for individual states, and some states may screen for disorders that are not included on this list of recommended disorders. Prior to this, babies born in different states had received different levels of screening. On April 24, 2008, President George W. Bush signed into law the Newborn Screening Saves Lives Act of 2007. This act was enacted to increase awareness among parents, health professionals, and the public on testing newborns to identify certain disorders. It also sought to improve, expand, and enhance current newborn screening programs at the state level.[citation needed]
Inclusion of disorders
Newborn screening programs initially used screening criteria based largely on criteria established by JMG Wilson and F. Jungner in 1968.[6] Although not specifically about newborn population screening programs, their publication, Principles and practice of screening for disease proposed ten criteria that screening programs should meet before being used as a public health measure. Newborn screening programs are administered in each jurisdiction, with additions and removals from the panel typically reviewed by a panel of experts. The four criteria from the publication that were relied upon when making decisions for early newborn screening programs were: [citation needed]
having an acceptable treatment protocol in place that changes the outcome for patients diagnosed early with the disease
an understanding of the condition's natural history
an understanding about who will be treated as a patient
a screening test that is reliable for both affected and unaffected patients and is acceptable to the public[7]
As diagnostic techniques have progressed, debates have arisen as to how screening programs should adapt. Tandem mass spectrometry has greatly expanded the potential number of diseases that can be detected, even without satisfying all of the other criteria used for making screening decisions.[7][8]Duchenne muscular dystrophy is a disease that has been added to screening programs in several jurisdictions around the world, despite the lack of evidence as to whether early detection improves the clinical outcome for a patient.[7]
Newborn screening is intended as a public health program to identify infants with treatable conditions before they present clinically, or suffer irreversible damage. Phenylketonuria (PKU) was the first disorder targeted for newborn screening, being implemented in a small number of hospitals and quickly expanding across the United States and the rest of the world.[9] After the success of newborn screening for PKU (39 infants were identified and treated in the first two years of screening, with no false negative results), Guthrie and others looked for other disorders that could be identified and treated in infants, eventually developing bacterial inhibition assays to identify classic galactosemia and maple syrup urine disease.[9][10]
Newborn screening has expanded since the introduction of PKU testing in the 1960s, but can vary greatly between countries. In 2011, the United States screened for 54 conditions, Germany for 12, the United Kingdom for 2 (PKU and medium chain acyl-CoA dehydrogenase deficiency (MCADD)), while France and Hong Kong only screened for one condition (PKU and congenital hypothyroidism, respectively).[11] The conditions included in newborn screening programs around the world vary greatly, based on the legal requirements for screening programs, prevalence of certain diseases within a population, political pressure, and the availability of resources for both testing and follow-up of identified patients.[citation needed]
Congenital Disorders of Amino Acid Metabolism
Newborn screening originated with an amino acid disorder, phenylketonuria (PKU), which can be easily treated by dietary modifications, but causes severe Intellectual disability if not identified and treated early. Robert Guthrie introduced the newborn screening test for PKU in the early 1960s.[12] With the knowledge that PKU could be detected before symptoms were evident, and treatment initiated, screening was quickly adopted around the world. Ireland was the first country in the world to introduce a nationwide screening programme in February 1966,[13] Austria started screening the same year[14] and England in 1968.[15]
With the advent of tandem mass spectrometry as a screening tool, several fatty acid oxidation disorders were targeted for inclusion in newborn screening programs. Medium chain acyl-CoA dehydrogenase deficiency (MCADD), which had been implicated in several cases of sudden infant death syndrome[16][17][18] was one of the first conditions targeted for inclusion. MCADD was the first condition added when the United Kingdom expanded their screening program from PKU only.[11] Population based studies in Germany, the United States and Australia put the combined incidence of fatty acid oxidation disorders at 1:9300 among Caucasians. The United States screens for all known fatty acid oxidation disorders, either as primary or secondary targets, while other countries screen for a subset of these.[19]
The introduction of screening for fatty acid oxidation disorders has been shown to have reduced morbidity and mortality associated with the conditions, particularly MCADD. An Australian study found a 74% reduction in episodes of severe metabolic decompensation or death among individuals identified by newborn screening as having MCADD versus those who presented clinically prior to screening. Studies in the Netherlands and United Kingdom found improvements in outcome at a reduced cost when infants were identified before presenting clinically.[19]
Newborn screening programs have also expanded the information base available about some rare conditions. Prior to its inclusion in newborn screening, short-chain acyl-CoA dehydrogenase deficiency (SCADD) was thought to be life-threatening. Most patients identified via newborn screening as having this enzyme deficiency were asymptomatic, to the extent that SCADD was removed from screening panels in a number of regions. Without the cohort of patients identified by newborn screening, this clinical phenotype would likely not have been identified.[19]
Endocrinopathies
The most commonly included disorders of the endocrine system are congenital hypothyroidism (CH) and congenital adrenal hyperplasia (CAH).[20] Testing for both disorders can be done using blood samples collected on the standard newborn screening card. Screening for CH is done by measuring thyroxin (T4), thyrotropin (TSH) or a combination of both analytes. Elevated 17α-hydroxyprogesterone (17α-OHP) is the primary marker used when screening for CAH, most commonly done using enzyme-linked immunosorbant assays, with many programs using a second tier tandem mass spectrometry test to reduce the number of false positive results.[20] Careful analysis of screening results for CAH may also identify cases of congenital adrenal hypoplasia, which presents with extremely low levels of 17α-OHP.[20] When the immunoassay method is utilized as a screening method for quantifying 17α-OHP in dried blood spots, it exhibits a significant rate of false positive results. As per the clinical practice guideline issued by the Endocrine Society in 2018, employing LC-MS/MS to measure 17α-OHP and other adrenal steroid hormones (such as 21-deoxycortisol and androstenedione) is recommended as a supplementary screening approach to enhance the accuracy of positive predictions.[21]
CH was added to many newborn screening programs in the 1970s, often as the second condition included after PKU. The most common cause of CH is dysgenesis of the thyroid gland After many years of newborn screening, the incidence of CH worldwide had been estimated at 1:3600 births, with no obvious increases in specific ethnic groups. Recent data from certain regions have shown an increase, with New York reporting an incidence of 1:1700. Reasons for the apparent increase in incidence have been studied, but no explanation has been found.[20]
Classic CAH, the disorder targeted by newborn screening programs, is caused by a deficiency of the enzyme steroid 21-hydroxylase and comes in two forms – simple virilizing and a salt-wasting form. The incidence of CAH can vary greatly between populations. The highest reported incidence rates are among the Yupic Eskimos of Alaska (1:280) and on the French island of Réunion (1:2100).[20]
Hemoglobinopathies
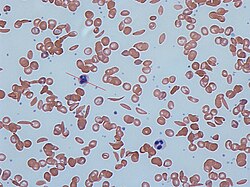
Sickle cells in human blood: both normal red blood cells and sickle-shaped cells are present
Any condition that results in the production of abnormal hemoglobin is included under the broad category of hemoglobinopathies. Worldwide, it is estimated that 7% of the population may carry a hemoglobinopathy with clinical significance.[22] The most well known condition in this group is sickle cell disease.[22] Newborn screening for a large number of hemoglobinopathies is done by detecting abnormal patterns using isoelectric focusing, which can detect many different types of abnormal hemoglobins.[22] In the United States, newborn screening for sickle cell disease was recommended for all infants in 1987, however it was not implemented in all 50 states until 2006.[22]
Early identification of individuals with sickle cell disease and other hemoglobinopathies allows treatment to be initiated in a timely fashion. Penicillin has been used in children with sickle cell disease, and blood transfusions are used for patients identified with severe thalassemia.[22]
Organic acidemias
Most jurisdictions did not start screening for any of the organic acidemias before tandem mass spectrometry significantly expanded the list of disorders detectable by newborn screening. Quebec has run a voluntary second-tier screening program since 1971 using urine samples collected at three weeks of age to screen for an expanded list of organic acidemias using a thin layer chromatography method.[23] Newborn screening using tandem mass spectrometry can detect several organic acidemias, including propionic acidemia, methylmalonic acidemia and isovaleric acidemia.[citation needed]
Cystic fibrosis
Cystic fibrosis (CF) was first added to newborn screening programs in New Zealand and regions of Australia in 1981, by measuring immunoreactive trypsinogen (IRT) in dried blood spots.[24] After the CFTR gene was identified, Australia introduced a two tier testing program to reduce the number of false positives. Samples with an elevated IRT value were then analyzed with molecular methods to identify the presence of disease causing mutations before being reported back to parents and health care providers.[25] CF is included in the core panel of conditions recommended for inclusion in all 50 states, Texas was the last state to implement their screening program for CF in 2010.[26] Alberta was the first Canadian province to implement CF screening in 2007.[27] Quebec, New Brunswick, Nova Scotia, Newfoundland and Prince Edward Island do not include CF in their screening programs.[28] The United Kingdom as well as many European Union countries screen for CF as well.[28] Switzerland is one of the latest countries to add CF to their newborn screening menu, doing so in January 2011.[24]
Urea cycle disorders
Disorders of the distal urea cycle, such as citrullinemia, argininosuccinic aciduria and argininemia are included in newborn screening programs in many jurisdictions that using tandem mass spectrometry to identify key amino acids. Proximal urea cycle defects, such as ornithine transcarbamylase deficiency and carbamoyl phosphate synthetase deficiency are not included in newborn screening panels because they are not reliably detected using current technology, and also because severely affected infants will present with clinical symptoms before newborn screening results are available. Some regions claim to screen for HHH syndrome (hyperammonemia, hyperornithinemia, homocitrullinuria) based on the detection of elevated ornithine levels in the newborn screening dried blood spot, but other sources have shown that affected individuals do not have elevated ornithine at birth.[29]
Lysosomal storage disorders
Lysosomal storage disorders are not included in newborn screening programs with high frequency. As a group, they are heterogenous, with screening only being feasible for a small fraction of the approximately 40 identified disorders. The arguments for their inclusion in newborn screening programs center around the advantage of early treatment (when treatment is available), avoiding a diagnostic odyssey for families and providing information for family planning to couples who have an affected child.[30] The arguments against including these disorders, as a group or individually center around the difficulties with reliably identifying individuals who will be affected with a severe form of the disorder, the relatively unproven nature of the treatment methods, and the high cost / high risk associated with some treatment options.[30]
New York State started a pilot study to screen for Krabbe disease in 2006, largely due to the efforts of Jim Kelly, whose son, Hunter, was affected with the disease.[31] A pilot screening program for four lysosomal storage diseases (Gaucher disease, Pompe disease, Fabry disease and Niemann-Pick disease was undertaken using anonymised dried blood spots was completed in Austria in 2010. Their data showed an increased incidence from what was expected in the population, and also a number of late onset forms of disease, which are not typically the target for newborn screening programs.[32]
Hearing loss
Undiagnosed hearing loss in a child can have serious effects on many developmental areas, including language, social interactions, emotions, cognitive ability, academic performance and vocational skills, any combination of which can have negative impacts on the quality of life.[33] The serious impacts of a late diagnosis, combined with the high incidence (estimated at 1 - 3 per 1000 live births, and as high as 4% for neonatal intensive care unit patients) have been the driving forces behind screening programs designed to identify infants with hearing loss as early as possible. Early identification allows these patients and their families to access the necessary resources to help them maximize their developmental outcomes.[33]
Newborn Hearing Screening
Newborn hearing testing is done at the bedside using transiently evoked otoacoustic emissions, automated auditory brainstem responses, or a combination of both techniques. Hearing screening programs have found the initial testing to cost between $10.20 and $23.37 per baby, depending on the technology used.[33] As these are screening tests only, false positive results will occur. False positive results could be due to user error, a fussy baby, environmental noise in the testing room, or fluid or congestion in the outer/middle ear of the baby. A review of hearing screening programs found varied initial referral rates (screen positive results) from 0.6% to 16.7%. The highest overall incidence of hearing loss detection was 0.517%.[33] A significant proportion of screen positive infants were lost to follow-up before a diagnosis could be confirmed or ruled out in all screening programs.[33]
Congenital heart defects
In some cases, critical congenital heart defects (CCHD) are not identified by prenatal ultrasound or postnatal physical examination. Pulse oximetry has been recently added as a bedside screening test for CCHD[34] at 24 to 48 hours after birth. However, not all heart problems can be detected by this method, which relies only on blood oxygen levels.
When a baby tests positive, urgent subsequent examination, such as echocardiography, is undergone to determine the cause of low oxygen levels. Babies diagnosed with CCHD are then seen by cardiologists.[citation needed]
Severe combined immunodeficiency
Severe combined immunodeficiency (SCID) caused by T-cell deficiency is a disorder that was recently added to newborn screening programs in some regions of the United States. Wisconsin was the first state to add SCID to their mandatory screening panel in 2008, and it was recommended for inclusion in all states' panels in 2010. Since December 2018 all US states perform SCID screening.[35] As the first country in Europe, Norway started nationwide SCID screening January 2018.[36][37] Identification of infants with SCID is done by detecting T-cell receptor excision circles (TRECs) using real-time polymerase chain reaction (qPCR). TRECs are decreased in infants affected with SCID.[38]
SCID has not been added to newborn screening in a wide scale for several reasons. It requires technology that is not currently used in most newborn screening labs, as PCR is not used for any other assays included in screening programs. Follow-up and treatment of affected infants also requires skilled immunologists, which may not be available in all regions. Treatment for SCID is a stem cell transplant, which cannot be done in all centers.[38]
Other conditions
Duchenne muscular dystrophy (DMD) is an X-linked disorder caused by defective production of dystrophin. Many jurisdictions around the world have screened for, or attempted to screen for DMD using elevated levels of creatine kinase measured in dried blood spots. Because universal newborn screening for DMD has not been undertaken, affected individuals often have a significant delay in diagnosis. As treatment options for DMD become more and more effective, interest in adding a newborn screening test increases. At various times since 1978, DMD has been included (often as a pilot study on a small subset of the population) in newborn screening programs in Edinburgh, Germany, Canada, France, Wales, Cyprus, Belgium and the United States. In 2012, Belgium was the only country that continued to screen for DMD using creatine kinase levels.[39]
As treatments improve, newborn screening becomes a possibility for disorders that could benefit from early intervention, but none was previously available. Adrenoleukodystrophy (ALD), a peroxisomal disease that has a variable clinical presentation is one of the disorders that has become a target for those seeking to identify patients early. ALD can present in several different forms, some of which do not present until adulthood, making it a difficult choice for countries to add to screening programs. The most successful treatment option is a stem cell transplant, a procedure that carries a significant risk.[40]
Techniques
Sample collection
Newborn screening tests are most commonly done from whole blood samples collected on specially designed filter paper, originally designed by Robert Guthrie. The filter paper is often attached to a form containing required information about the infant and parents. This includes date and time of birth, date and time of sample collection, the infant's weight and gestational age. The form will also have information about whether the baby has had a blood transfusion and any additional nutrition the baby may have received (total parenteral nutrition). Most newborn screening cards also include contact information for the infant's physician in cases where follow up screening or treatment is needed. The Canadian province of Quebec performs newborn screening on whole blood samples collected as in most other jurisdictions, and also runs a voluntary urine screening program where parents collect a sample at 21 days of age and submit it to a provincial laboratory for an additional panel of conditions.[41][23]
Newborn screening samples are collected from the infant between 24 hours and 7 days after birth, and it is recommended that the infant has fed at least once. Individual jurisdictions will often have more specific requirements, with some states accepting samples collected at 12 hours, and others recommending to wait until 48 hours of life or later. Each laboratory will have its own criteria on when a sample is acceptable, or if another would need to be collected. Samples can be collected at the hospital, or by midwives. Samples are transported daily to the laboratory responsible for testing. In the United States and Canada, newborn screening is mandatory, with an option for parents to opt out of the screening in writing if they desire. In many regions, NBS is mandatory, with an option for parents to opt out in writing if they choose not to have their infant screened.[42] In most of Europe, newborn screening is done with the consent of the parents. Proponents of mandatory screening claim that the test is for the benefit of the child, and that parents should not be able to opt out on their behalf. In regions that favour informed consent for the procedure, they report no increase in costs, no decrease in the number of children screened and no cases of included diseases in children who did not undergo screening.[43]
As of 2023, numerous initiatives using next generation sequencing (NGS) have been announced worldwide including the Genomic Uniform-screening Against Rare Diseases in All Newborns (GUARDIAN study), BeginNGS and Early Check in the USA, BabyScreen+ in Australia, Generation Study by Genomics England,[44] and Screen4Care,[45] Baby Detect in Belgium[46] and PERIGENOMED in France.[47] In a 2023 survey of 14 European newborn screening programs, there was one pan-European research study with 2 pilot trials planned in Germany (NEW_LIVES)[48] and Italy, the others included three initiatives in Italy, three in the Netherlands, two in Spain, one in Belgium, one in England, one in Germany, one in Greece[49] and one in France. Of the 14 initiatives, 11 selected a single NGS approach for their studies: 6 initiatives planned to use only whole genome sequencing (WGS) as a first-tier test for NBS, including one also testing parents using whole exome sequencing (WES) to facilitate filtering variants, 3 initiatives use classical NGS gene panels, 2 initiatives will be using WES and 2 initiatives will use a mixed approach: one comparing WES and Whole genome sequencing (WGS) and one comparing WES, WGS, and classical NGS. gene panels.[47]
Reporting results
The goal is to report the results within a short period of time. If screens are normal, a paper report is sent to the submitting hospital and parents rarely hear about it. If an abnormality is detected, employees of the agency, usually nurses, begin to try to reach the physician, hospital, and/or nursery by telephone. They are persistent until they can arrange an evaluation of the infant by an appropriate specialist physician (depending on the disease). The specialist will attempt to confirm the diagnosis by repeating the tests by a different method or laboratory, or by performing other corroboratory or disproving tests. The confirmatory test varies depending on the positive results on the initial screen. Confirmatory testing can include analyte specific assays to confirm any elevations detected, functional studies to determine enzyme activity, and genetic testing to identify disease-causing mutations. In some cases, a positive newborn screen can also trigger testing on other family members, such as siblings who did not undergo newborn screening for the same condition or the baby's mother, as some maternal conditions can be identified through results on the baby's newborn screen. Depending on the likelihood of the diagnosis and the risk of delay, the specialist will initiate treatment and provide information to the family. Performance of the program is reviewed regularly and strenuous efforts are made to maintain a system that catches every infant with these diagnoses. Guidelines for newborn screening and follow up have been published by the American Academy of Pediatrics[50] and the American College of Medical Genetics.[51]
Laboratory performance
Newborn screening programs participate in quality control programs as in any other laboratory, with some notable exceptions. Much of the success of newborn screening programs is dependent on the filter paper used for the collection of the samples. Initial studies using Robert Guthrie's test for PKU reported high false positive rates that were attributed to a poorly selected type of filter paper.[52] This source of variation has been eliminated in most newborn screening programs through standardization of approved sources of filter paper for use in newborn screening programs. In most regions, the newborn screening card (which contains demographic information as well as attached filter paper for blood collection) is supplied by the organization carrying out the testing, to remove variations from this source.[52]
Society and culture
Controversy
Newborn screening tests have become a subject of political controversy in the last decade[clarification needed]. Lawsuits, media attention, and advocacy groups have surfaced a number of different, and possibly countervailing, positions on the use of screening tests. Some have asked for government mandates to widen the extent of the screening to find detectable and treatable birth defects. Others have opposed mandatory screening concerned that effective follow-up and treatment may not be available, or that false positive screening tests may cause harm to infants and their families. Others have learned that government agencies were often secretly storing the results in databases for future genetic research, often without consent of the parents nor limits on how the data could be used in the future [citation needed]. In the UK a campaign called the Newborn Screening Collaborative, 17 small rare disease organisations including Genetic Alliance UK, have joined together to raise awareness surrounding this issue and promote the positives of early diagnosis.[citation needed]
Increasing mandatory tests in California
Many rare diseases have not historically been tested for or testing that has been available has not been mandatory. One such disease is glutaric acidemia type I, a neurometabolic disease present in approximately 1 out of every 100,000 live births.[53] A short-term California testing pilot project in 2003 and 2004 demonstrated the cost of forgoing rare disease testing on newborns. While both Zachary Wyvill and Zachary Black were both born with the same disease during the pilot program, Wyvill's birth hospital tested only for four state-mandated diseases while Black was born at a hospital participating in the pilot program. Wyvill's disease went undetected for over six months during which irreversible damage occurred but Black's disease was treated with diet and vitamin supplements.[54] Both sets of parents became advocates for expanded neonatal testing and testified in favor of expanding tandem mass spectrometry (MS/MS) testing of newborns for rare diseases. By August, 2004, the California state budget law had passed requiring the use of tandem mass spectroscopy to test for more than 30 genetic illnesses and provided funding.[55] California now mandates newborn screening for all infants and tests for 80 congenital and genetic disorders.[56]
Government budgetary limitations
Instituting MS/MS screening often requires a sizable up front expenditure. When states choose to run their own programs the initial costs for equipment, training and new staff can be significant. Moreover, MS/MS gives only the screening result and not the confirmatory result. The same has to be further done by higher technologies or procedure like GC/MS[clarification needed], Enzyme Assays or DNA Tests. This in effect adds more cost burden and makes physicians lose precious time.[according to whom?] To avoid at least a portion of the up front costs, some states such as Mississippi have chosen to contract with private labs for expanded screening. Others have chosen to form Regional Partnerships sharing both costs and resources.[citation needed]
But for many states, screening has become an integrated part of the department of health which can not or will not be easily replaced. Thus the initial expenditures can be difficult for states with tight budgets to justify. Screening fees have also increased in recent years as health care costs rise and as more states add MS/MS screening to their programs. (See Report of Summation of Fees Charged for Newborn Screening, 2001–2005) Dollars spent for these programs may reduce resources available to other potentially lifesaving programs. It was recommended[by whom?] in 2006 that one disorder, Short Chain Acyl-coenzymeA Dehydrogenase Deficiency, or SCAD, be eliminated from screening programs, due to a "spurious association between SCAD and symptoms.[57] However, other[when?] studies suggested that perhaps expanded screening is cost effective (see ACMG report page 94-95[dead link] and articles published in Pediatrics[58]'.[59] Advocates are quick to point out studies such as these when trying to convince state legislatures to mandate expanded screening.[citation needed]
Decreasing mandatory tests
Expanded newborn screening is also opposed by among some health care providers, who are concerned that effective follow-up and treatment may not be available, that false positive screening tests may cause harm, and issues of informed consent.[60] A recent study by Genetic Alliance and partners suggests that communication between health care providers and parents may be key in minimizing the potential harm when a false positive test occurs. The results from this study also reveal that parents found newborn screening to be a beneficial and necessary tool to prevent treatable diseases.[61] To address the false positive issue, researchers from the University of Maryland, Baltimore and Genetic Alliance established a check-list to assist health care providers communicate with parents about a screen-positive result.[62]
Secret genetic research
Controversy has also erupted in some countries over collection and storage of blood or DNA samples by government agencies during the routine newborn blood screen.[citation needed]
In the United States, it was revealed that Texas had collected and stored blood and DNA samples on millions of newborns without the parents' knowledge or consent. These samples were then used by the state for genetic experiments and to set up a database to catalog all of the samples/newborns. As of December2009[update], samples obtained without parents' consent between 2002 and 2009 were slated to be destroyed following the settlement of "a lawsuit filed by parents against the Texas Department of Health Services and Texas A&M; for secretly storing and doing research on newborn blood samples."[63]
A similar legal case was filed against the State of Minnesota. Over 1million newborn bloodspot samples were destroyed in 2011 "when the state's Supreme Court found that storage and use of blood spots beyond newborn screening panels was in violation of the state's genetic privacy laws.".[citation needed] Nearly US$1 million was required to be paid by the state for the attorney's fees of the 21 families who advanced the lawsuit. An advocacy group that has taken a position against research on newborn blood screening data without parental consent is the Citizens' Council for Health Freedom, who take the position that newborn health screening for "a specific set of newborn genetic conditions" is a very different matter than storing the data or those DNA samples indefinitely to "use them for genetic research without parental knowledge or consent."[citation needed]
Bioethics
As additional tests are discussed for addition to the panels, issues arise. Many question whether the expanded testing still falls under the requirements necessary to justify the additional tests.[64] Many of the new diseases being tested for are rare and have no known treatment, while some of the diseases need not be treated until later in life.[64] This raises more issues, such as: if there is no available treatment for the disease should we test for it at all? And if we do, what do we tell the families of those with children bearing one of the untreatable diseases?[65] Studies show that the rarer the disease is and the more diseases being tested for, the more likely the tests are to produce false-positives.[66] This is an issue because the newborn period is a crucial time for the parents to bond with the child, and it has been noted that ten percent of parents whose children were diagnosed with a false-positive still worried that their child was fragile and/or sickly even though they were not, potentially preventing the parent-child bond forming as it would have otherwise.[65] As a result, some parents may begin to opt out of having their newborns screened. Many parents are also concerned about what happens with their infant's blood samples after screening. The samples were originally taken to test for preventable diseases, but with the advance in genomic sequencing technologies many samples are being kept for DNA identification and research,[64][65] increasing the possibility that more children will be opted out of newborn screening from parents who see the kept samples as a form of research done on their child.[64]
See also
Euphenics– American molecular biologist (1925–2008)Pages displaying short descriptions of redirect targets
↑ Clague A, Thomas A (January 2002). "Neonatal biochemical screening for disease". Clinica Chimica Acta; International Journal of Clinical Chemistry. 315 (1–2): 99–110. doi:10.1016/S0009-8981(01)00716-1. PMID11728413.
↑ Wilson JM, Jungner YG (October 1968). "[Principles and practice of mass screening for disease]". Boletin de la Oficina Sanitaria Panamericana. Pan American Sanitary Bureau. 65 (4): 281–393. PMID4234760.
1 2 3 Ross LF (April 2006). "Screening for conditions that do not meet the Wilson and Jungner criteria: the case of Duchenne muscular dystrophy". American Journal of Medical Genetics. Part A. 140 (8): 914–922. doi:10.1002/ajmg.a.31165. PMID16528755. S2CID24612331.
↑ Korman SH, Gutman A, Brooks R, Sinnathamby T, Gregersen N, Andresen BS (June 2004). "Homozygosity for a severe novel medium-chain acyl-CoA dehydrogenase (MCAD) mutation IVS3-1G > C that leads to introduction of a premature termination codon by complete missplicing of the MCAD mRNA and is associated with phenotypic diversity ranging from sudden neonatal death to asymptomatic status". Molecular Genetics and Metabolism. 82 (2): 121–129. doi:10.1016/j.ymgme.2004.03.002. PMID15171999.
↑ Gregersen N, Winter V, Jensen PK, Holmskov A, Kølvraa S, Andresen BS, etal. (January 1995). "Prenatal diagnosis of medium-chain acyl-CoA dehydrogenase (MCAD) deficiency in a family with a previous fatal case of sudden unexpected death in childhood". Prenatal Diagnosis. 15 (1): 82–86. doi:10.1002/pd.1970150118. PMID7740006. S2CID24295134.
1 2 3 Lindner M, Hoffmann GF, Matern D (October 2010). "Newborn screening for disorders of fatty-acid oxidation: experience and recommendations from an expert meeting". Journal of Inherited Metabolic Disease. 33 (5): 521–526. doi:10.1007/s10545-010-9076-8. PMID20373143. S2CID1794910.
1 2 3 4 5 Pass KA, Neto EC (December 2009). "Update: newborn screening for endocrinopathies". Endocrinology and Metabolism Clinics of North America. 38 (4): 827–837. doi:10.1016/j.ecl.2009.08.005. PMID19944295.
↑ Mu D, Sun D, Qian X, Ma X, Qiu L, Cheng X, etal. (January 2024). "Steroid profiling in adrenal disease". Clinica Chimica Acta; International Journal of Clinical Chemistry. 553 117749. doi:10.1016/j.cca.2023.117749. PMID38169194. S2CID266721414.
↑ Mechtler TP, Stary S, Metz TF, De Jesús VR, Greber-Platzer S, Pollak A, etal. (January 2012). "Neonatal screening for lysosomal storage disorders: feasibility and incidence from a nationwide study in Austria". Lancet. 379 (9813): 335–341. doi:10.1016/S0140-6736(11)61266-X. PMID22133539. S2CID23650785.
1 2 3 4 5 6 Papacharalampous GX, Nikolopoulos TP, Davilis DI, Xenellis IE, Korres SG (October 2011). "Universal newborn hearing screening, a revolutionary diagnosis of deafness: real benefits and limitations". European Archives of Oto-Rhino-Laryngology. 268 (10): 1399–1406. doi:10.1007/s00405-011-1672-1. PMID21698417. S2CID20647009.
1 2 Thangaratinam S, Brown K, Zamora J, Khan KS, Ewer AK (June 2012). "Pulse oximetry screening for critical congenital heart defects in asymptomatic newborn babies: a systematic review and meta-analysis". Lancet. 379 (9835): 2459–2464. doi:10.1016/S0140-6736(12)60107-X. PMID22554860. S2CID19949842.
↑ Raymond GV, Jones RO, Moser AB (2007). "Newborn screening for adrenoleukodystrophy: implications for therapy". Molecular Diagnosis & Therapy. 11 (6): 381–384. doi:10.1007/BF03256261. PMID18078355. S2CID21323198.
1 2 De Jesús VR, Mei JV, Bell CJ, Hannon WH (April 2010). "Improving and assuring newborn screening laboratory quality worldwide: 30-year experience at the Centers for Disease Control and Prevention". Seminars in Perinatology. 34 (2): 125–133. doi:10.1053/j.semperi.2009.12.003. PMID20207262.
↑ Schulze A, Lindner M, Kohlmüller D, Olgemöller K, Mayatepek E, Hoffmann GF (June 2003). "Expanded newborn screening for inborn errors of metabolism by electrospray ionization-tandem mass spectrometry: results, outcome, and implications". Pediatrics. 111 (6 Pt 1): 1399–1406. doi:10.1542/peds.111.6.1399. PMID12777559.
↑ Schoen EJ, Baker JC, Colby CJ, To TT (October 2002). "Cost-benefit analysis of universal tandem mass spectrometry for newborn screening". Pediatrics. 110 (4): 781–786. doi:10.1542/peds.110.4.781. PMID12359795.
1 2 3 Clayton EW (2003). Newborn Genetic Screening. Contemporary Issues in Bioethics: Thomas Wadsworth. pp.248–251. ISBN978-0-495-00673-2.
↑ Tarini BA, Christakis DA, Welch HG (August 2006). "State newborn screening in the tandem mass spectrometry era: more tests, more false-positive results". Pediatrics. 118 (2): 448–456. doi:10.1542/peds.2005-2026. PMID16882794. S2CID28070141.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.