Related Research Articles
Glucose-6-phosphate dehydrogenase deficiency (G6PDD), also known as favism, is the most common enzyme deficiency anemia worldwide. It is an inborn error of metabolism that predisposes to red blood cell breakdown. Most of the time, those who are affected have no symptoms. Following a specific trigger, symptoms such as yellowish skin, dark urine, shortness of breath, and feeling tired may develop. Complications can include anemia and newborn jaundice. Some people never have symptoms.

Galactosemia is a rare genetic metabolic disorder that affects an individual's ability to metabolize the sugar galactose properly. Galactosemia follows an autosomal recessive mode of inheritance that confers a deficiency in an enzyme responsible for adequate galactose degradation.

In biochemistry, a transferase is any one of a class of enzymes that catalyse the transfer of specific functional groups from one molecule to another. They are involved in hundreds of different biochemical pathways throughout biology, and are integral to some of life's most important processes.

Newborn screening (NBS) is a public health program of screening in infants shortly after birth for conditions that are treatable, but not clinically evident in the newborn period. The goal is to identify infants at risk for these conditions early enough to confirm the diagnosis and provide intervention that will alter the clinical course of the disease and prevent or ameliorate the clinical manifestations. NBS started with the discovery that the amino acid disorder phenylketonuria (PKU) could be treated by dietary adjustment, and that early intervention was required for the best outcome. Infants with PKU appear normal at birth, but are unable to metabolize the essential amino acid phenylalanine, resulting in irreversible intellectual disability. In the 1960s, Robert Guthrie developed a simple method using a bacterial inhibition assay that could detect high levels of phenylalanine in blood shortly after a baby was born. Guthrie also pioneered the collection of blood on filter paper which could be easily transported, recognizing the need for a simple system if the screening was going to be done on a large scale. Newborn screening around the world is still done using similar filter paper. NBS was first introduced as a public health program in the United States in the early 1960s, and has expanded to countries around the world.
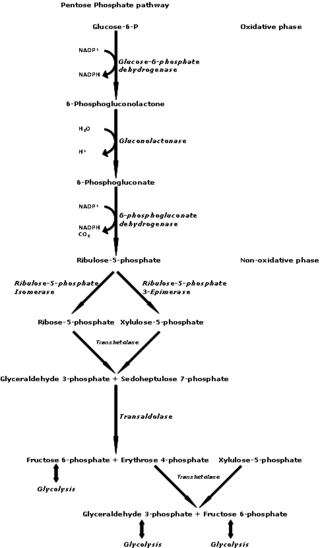
The pentose phosphate pathway is a metabolic pathway parallel to glycolysis. It generates NADPH and pentoses as well as ribose 5-phosphate, a precursor for the synthesis of nucleotides. While the pentose phosphate pathway does involve oxidation of glucose, its primary role is anabolic rather than catabolic. The pathway is especially important in red blood cells (erythrocytes). The reactions of the pathway were elucidated in the early 1950s by Bernard Horecker and co-workers.

Galactose-1-phosphate uridyltransferase is an enzyme responsible for converting ingested galactose to glucose.
Glucose-6-phosphate isomerase (GPI), alternatively known as phosphoglucose isomerase/phosphoglucoisomerase (PGI) or phosphohexose isomerase (PHI), is an enzyme that in humans is encoded by the GPI gene on chromosome 19. This gene encodes a member of the glucose phosphate isomerase protein family. The encoded protein has been identified as a moonlighting protein based on its ability to perform mechanistically distinct functions. In the cytoplasm, the gene product functions as a glycolytic enzyme that interconverts glucose-6-phosphate (G6P) and fructose-6-phosphate (F6P). Extracellularly, the encoded protein functions as a neurotrophic factor that promotes survival of skeletal motor neurons and sensory neurons, and as a lymphokine that induces immunoglobulin secretion. The encoded protein is also referred to as autocrine motility factor (AMF) based on an additional function as a tumor-secreted cytokine and angiogenic factor. Defects in this gene are the cause of nonspherocytic hemolytic anemia, and a severe enzyme deficiency can be associated with hydrops fetalis, immediate neonatal death and neurological impairment. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Jan 2014]

Glucose-6-phosphate dehydrogenase (G6PD or G6PDH) (EC 1.1.1.49) is a cytosolic enzyme that catalyzes the chemical reaction

Galactokinase deficiency is an autosomal recessive metabolic disorder marked by an accumulation of galactose and galactitol secondary to the decreased conversion of galactose to galactose-1-phosphate by galactokinase. The disorder is caused by mutations in the GALK1 gene, located on chromosome 17q24. Galactokinase catalyzes the first step of galactose phosphorylation in the Leloir pathway of intermediate metabolism. Galactokinase deficiency is one of the three inborn errors of metabolism that lead to hypergalactosemia. The disorder is inherited as an autosomal recessive trait. Unlike classic galactosemia, which is caused by deficiency of galactose-1-phosphate uridyltransferase, galactokinase deficiency does not present with severe manifestations in early infancy. Its major clinical symptom is the development of cataracts during the first weeks or months of life, as a result of the accumulation, in the lens, of galactitol, a product of an alternative route of galactose utilization. The development of early cataracts in homozygous affected infants is fully preventable through early diagnosis and treatment with a galactose-restricted diet. Some studies have suggested that, depending on milk consumption later in life, heterozygous carriers of galactokinase deficiency may be prone to presenile cataracts at 20–50 years of age.

Galactose epimerase deficiency, also known as GALE deficiency, Galactosemia III and UDP-galactose-4-epimerase deficiency, is a rare, autosomal recessive form of galactosemia associated with a deficiency of the enzyme galactose epimerase.
Galactose-1-phosphate uridylyltransferase deficiency(classic galactosemia) is the most common type of galactosemia, an inborn error of galactose metabolism, caused by a deficiency of the enzyme galactose-1-phosphate uridylyltransferase. It is an autosomal recessive metabolic disorder that can cause liver disease and death if untreated. Treatment of galactosemia is most successful if initiated early and includes dietary restriction of lactose intake. Because early intervention is key, galactosemia is included in newborn screening programs in many areas. On initial screening, which often involves measuring the concentration of galactose in blood, classic galactosemia may be indistinguishable from other inborn errors of galactose metabolism, including galactokinase deficiency and galactose epimerase deficiency. Further analysis of metabolites and enzyme activities are needed to identify the specific metabolic error.
The enzyme UDP-glucose 4-epimerase, also known as UDP-galactose 4-epimerase or GALE, is a homodimeric epimerase found in bacterial, fungal, plant, and mammalian cells. This enzyme performs the final step in the Leloir pathway of galactose metabolism, catalyzing the reversible conversion of UDP-galactose to UDP-glucose. GALE tightly binds nicotinamide adenine dinucleotide (NAD+), a co-factor required for catalytic activity.

GDH/6PGL endoplasmic bifunctional protein is a protein that in humans is encoded by the H6PD gene.

Inborn errors of carbohydrate metabolism are inborn error of metabolism that affect the catabolism and anabolism of carbohydrates.

6-Phosphogluconate dehydrogenase deficiency, or partial deficiency, is an autosomal hereditary disease characterized by abnormally low levels of 6-phosphogluconate dehydrogenase (6PGD), a metabolic enzyme involved in the Pentose phosphate pathway. It is very important in the metabolism of red blood cells (erythrocytes). 6PDG deficiency affects less than 1% of the population, and studies suggest that there may be race variant involved in many of the reported cases. Although it is similar, 6PDG deficiency is not linked to glucose-6-phosphate dehydrogenase (G6PD) deficiency, as they are located on different chromosomes. However, a few people have had both of these metabolic diseases.
Ernest Beutler was a German-born American hematologist and biomedical scientist. He made important discoveries about the causes of a number of diseases, including anemias, Gaucher disease, disorders of iron metabolism and Tay–Sachs disease. He was also among the first scientists to identify X-inactivation as the genetic basis of tissue mosaicism in female mammals, and pioneered a number of medical treatments, including bone marrow transplantation techniques. Beutler was the Chairman of Medicine at the City of Hope Medical Center in Duarte, CA from 1959 until 1979 and served as a Professor, then Chairman, of the Department of Molecular and Experimental Medicine at The Scripps Research Institute in La Jolla, California from 1979 until 2008.
A broad classification for genetic disorders that result from an inability of the body to produce or utilize an enzyme or transport protein that is required to oxidize fatty acids. They are an inborn error of lipid metabolism, and when it affects the muscles also a metabolic myopathy.
Galactolysis refers to the catabolism of galactose.

Duarte galactosemia is an inherited condition associated with diminished ability to metabolize galactose due to a partial deficiency of the enzyme galactose-1-phosphate uridylyltransferase. DG differs from classic galactosemia in that patients with Duarte galactosemia have partial GALT deficiency whereas patients with classic galactosemia have complete, or almost complete, GALT deficiency. Duarte galactosemia (DG) is much more common than classic galactosemia, and is estimated to affect close to one in 4,000 infants born in the United States. Historically, most healthcare professionals have considered DG to be clinically mild based on pilot studies and anecdotal experience, and in 2019 a large study confirmed that children with DG are not at increased risk for developmental problems relative to children who do not have DG. Due to regional variations in newborn screening (NBS) protocols, some infants with DG are identified by NBS but others are not.
References
- ↑ Markić J, Krzelj V, Markotić A, et al. (August 2006). "High incidence of glucose-6-phosphate dehydrogenase deficiency in Croatian island isolate: example from Vis island, Croatia". Croat. Med. J. 47 (4): 556–70. PMC 2080441 . PMID 16909453. Archived from the original on 2016-06-23. Retrieved 2008-08-14.
- ↑ Beutler E, Baluda MC. A simple spot screening test for galactosemia. J Lab Clin Med 1966;68:137-141.
- ↑ Beutler E, Baluda M, Donnell GE. A new method for the detection of galactosemia and its carrier state. J Lab Clin Med 1964;64:695-705.
- ↑ Beutler E, Mitchell M. New rapid for the estimation of red cell galactose-1-phosphate uridyl transferase activity. J Lab Clin Med 1968;72:527-532.
- ↑ Fujimoto A, Okano Y, Miyagi T, Isshiki G, Oura T (1999). "Mass screening of galactosemia: improved Beutler Test using automated quantitative fluorescence assay". Southeast Asian J. Trop. Med. Public Health. 30 (Suppl 2): 69. PMID 11400789.
- ↑ Fujimoto A, Okano Y, Miyagi T, Isshiki G, Oura T (June 2000). "Quantitative Beutler test for newborn mass screening of galactosemia using a fluorometric microplate reader". Clin. Chem. 46 (6 Pt 1): 806–10. doi: 10.1093/clinchem/46.6.806 . PMID 10839768. Archived from the original on 2020-08-16. Retrieved 2008-08-14.
- ↑ Kirimlidis S, Politis E, Drossos C, Scaloumbakas N, Papaioannou M (November 1965). "Glucose-6-phosphate-dehydrogenase deficiency in Greece. Study by using a modification of Beutler and Fair- banks spot test". Helv Paediatr Acta. 20 (5): 490–6. PMID 5895109.
- ↑ DERITIS L (April 1963). "The Fairbanks and Beutler Test for the Routine Detection of Erythrocyte Deficiency of Glucose-6-Phosphate Dehydrogenase". Riv Clin Pediatr (in Italian). 71: 242–4. PMID 14074804.