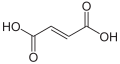
Argininosuccinic aciduria is an inherited disorder that causes the accumulation of argininosuccinic acid (also known as "ASA") in the blood and urine. Some patients may also have an elevation of ammonia, a toxic chemical, which can affect the nervous system. Argininosuccinic aciduria may become evident in the first few days of life because of high blood ammonia, or later in life presenting with "sparse" or "brittle" hair, developmental delay, and tremors.
An infant with argininosuccinic aciduria may seem lethargic or be unwilling to eat, have poorly controlled breathing rate or body temperature, experience seizures or unusual body movements, or go into a coma. Complications from argininosuccinic aciduria may include developmental delay and mental retardation. Progressive liver damage, skin lesions, and brittle hair may also be seen. Immediate treatment and lifelong management (following a strict diet and using appropriate supplements) may prevent many of these complications.
Occasionally, an individual may inherit a mild form of the disorder in which ammonia accumulates in the bloodstream only during periods of illness or other stress.
Genetics
Mutations in the ASLgene cause argininosuccinic aciduria. Argininosuccinic aciduria belongs to a class of genetic diseases called urea cycle disorders. The urea cycle is a sequence of reactions in the cells of the liver. It processes excess nitrogen, generated when protein is used by the body, to make a compound called urea that is excreted by the kidneys.[citation needed]
In argininosuccinic aciduria, the enzyme argininosuccinate lyase, involved in the conversion of arginino succinate to arginine within the urea cycle, is damaged or missing. The urea cycle cannot proceed normally, and nitrogen accumulates in the bloodstream in the form of ammonia. Ammonia is especially damaging to the nervous system, so argininosuccinic aciduria causes neurological problems as well as eventual damage to the liver.[citation needed]
This condition is inherited in an autosomal recessive pattern, which means two copies of the gene in each cell are altered. Most often, the parents of an individual with an autosomal recessive disorder are carriers of one copy of the altered gene but do not show signs and symptoms of the disorder.
Diagnosis
Diagnosis is based mainly on clinical findings and laboratory test results. Plasma concentrations of ammonia (>150 μmol/L) and citrulline (200-300 μmol/L) are elevated. Elevated levels of argininosuccinic acid (5-110 μmol/L) in the plasma or urine are diagnostic. Molecular genetic testing confirms diagnosis. Newborn screening for ASA is available in the U.S. and parts of Australia, and is considered in several European countries[citation needed]
Treatment
During an acute hyperammonemic episode, oral proteins must be avoided and intravenous (I.V.) lipids, glucose and insulin (if needed) should be given to promote anabolism. I.V. nitrogen scavenging therapy (with sodium benzoate and/or sodium phenylacetate) should normalize ammonia levels, but if unsuccessful, hemodialysis is recommended. Long-term management involves dietary protein restriction as well as arginine supplementation. In those with frequent episodes of metabolic decompensation or with hyperammonemia even when following a protein-restricted diet, daily oral nitrogen scavenging therapy may be successful. Orthotopic liver transplantation offers long-term relief of hyperammonemia but does not seem to sufficiently correct neurological complications. Arterial hypertension can be treated by restoring nitric oxide deficiency
Prognosis
Due to the rarity of the disease, it is hard to estimate mortality rates or life expectancy. One 2003 study which followed 88 cases receiving two different kinds of treatment found that very few persons lived beyond age 20 and none beyond age 30.[1]
Incidence
Argininosuccinic aciduria occurs in approximately 1 in 70,000 live births. Many patients can now be detected on the newborn screen if their blood citrulline is elevated.
Kleijer WJ, Garritsen VH, Linnebank M, Mooyer P, Huijmans JG, Mustonen A, Simola KO, Arslan-Kirchner M, Battini R, Briones P, Cardo E, Mandel H, Tschiedel E, Wanders RJ, Koch HG (2002). "Clinical, enzymatic, and molecular genetic characterization of a biochemical variant type of argininosuccinic aciduria: prenatal and postnatal diagnosis in five unrelated families". J Inherit Metab Dis. 25 (5): 399–410. doi:10.1023/A:1020108002877. PMID12408190. S2CID11129281.
Reid Sutton V, Pan Y, Davis EC, Craigen WJ (2003). "A mouse model of argininosuccinic aciduria: biochemical characterization". Mol Genet Metab. 78 (1): 11–6. doi:10.1016/S1096-7192(02)00206-8. PMID12559843.
Stadler S, Gempel K, Bieger I, Pontz BF, Gerbitz KD, Bauer MF, Hofmann S (2001). "Detection of neonatal argininosuccinate lyase deficiency by serum tandem mass spectrometry". J Inherit Metab Dis. 24 (3): 370–8. doi:10.1023/A:1010560704092. PMID11486903. S2CID11543156.
Wilcken B, Smith A, Brown DA (1980). "Urine screening for aminoacidopathies: is it beneficial? Results of a long-term follow-up of cases detected bny screening one million babies". J Pediatr. 97 (3): 492–7. doi:10.1016/S0022-3476(80)80216-2. PMID7411317.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.