
Riboflavin, also known as vitamin B2, is a vitamin found in food and sold as a dietary supplement. It is essential to the formation of two major coenzymes, flavin mononucleotide and flavin adenine dinucleotide. These coenzymes are involved in energy metabolism, cellular respiration, and antibody production, as well as normal growth and development. The coenzymes are also required for the metabolism of niacin, vitamin B6, and folate. Riboflavin is prescribed to treat corneal thinning, and taken orally, may reduce the incidence of migraine headaches in adults.
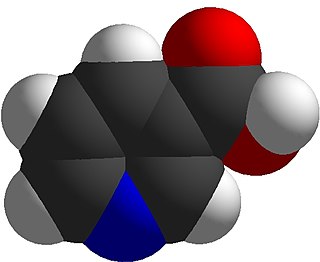
Niacin, also known as nicotinic acid, is an organic compound and a vitamer of vitamin B3, an essential human nutrient. It is produced by plants and animals from the amino acid tryptophan. Niacin is obtained in the diet from a variety of whole and processed foods, with highest contents in fortified packaged foods, meat, poultry, red fish such as tuna and salmon, lesser amounts in nuts, legumes and seeds. Niacin as a dietary supplement is used to treat pellagra, a disease caused by niacin deficiency. Signs and symptoms of pellagra include skin and mouth lesions, anemia, headaches, and tiredness. Many countries mandate its addition to wheat flour or other food grains, thereby reducing the risk of pellagra.
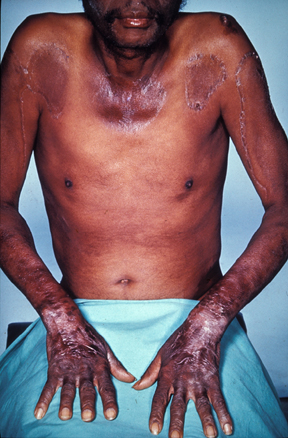
Pellagra is a disease caused by a lack of the vitamin niacin (vitamin B3). Symptoms include inflamed skin, diarrhea, dementia, and sores in the mouth. Areas of the skin exposed to friction and radiation are typically affected first. Over time affected skin may become darker, stiffen, peel, or bleed.

Cystinuria is an inherited autosomal recessive disease characterized by high concentrations of the amino acid cystine in the urine, leading to the formation of cystine stones in the kidneys, ureters, and bladder. It is a type of aminoaciduria. "Cystine", not "cysteine," is implicated in this disease; the former is a dimer of the latter.

Maple syrup urine disease (MSUD) is a rare, inherited metabolic disorder that affects the body's ability to metabolize amino acids due to a deficiency in the activity of the branched-chain alpha-ketoacid dehydrogenase (BCKAD) complex. It particularly affects the metabolism of amino acids—leucine, isoleucine, and valine. With MSUD, the body is not able to properly break down these amino acids, therefore leading to the amino acids to build up in urine and become toxic. The condition gets its name from the distinctive sweet odor of affected infants' urine and earwax due to the buildup of these amino acids.

Cystinosis is a lysosomal storage disease characterized by the abnormal accumulation of cystine, the oxidized dimer of the amino acid cysteine. It is a genetic disorder that follows an autosomal recessive inheritance pattern. It is a rare autosomal recessive disorder resulting from accumulation of free cystine in lysosomes, eventually leading to intracellular crystal formation throughout the body. Cystinosis is the most common cause of Fanconi syndrome in the pediatric age group. Fanconi syndrome occurs when the function of cells in renal tubules is impaired, leading to abnormal amounts of carbohydrates and amino acids in the urine, excessive urination, and low blood levels of potassium and phosphates.
Glutaric acidemia type 1 (GA1) is an inherited disorder in which the body is unable to completely break down the amino acids lysine, hydroxylysine and tryptophan. Excessive levels of their intermediate breakdown products can accumulate and cause damage to the brain, but particularly the basal ganglia, which are regions that help regulate movement. GA1 causes secondary carnitine deficiency, as glutaric acid, like other organic acids, is detoxified by carnitine. Mental retardation may occur.

Lysinuric protein intolerance (LPI) is an autosomal recessive metabolic disorder affecting amino acid transport. It is characterised by the body's inability to properly digest and use certain proteins. This condition leads to various metabolic complications and is typically diagnosed in infancy or early childhood.
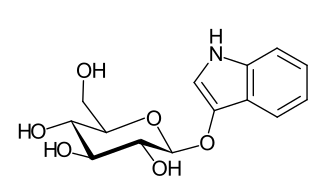
Indican is a colourless organic compound, soluble in water, naturally occurring in Indigofera plants. It is a precursor of indigo dye.

Blue diaper syndrome is a rare, autosomal recessive or X linked recessive metabolic disorder characterized in infants by bluish urine-stained diapers. It is also known as Drummond's syndrome, and hypercalcemia.

Aminoaciduria occurs when the urine contains abnormally high amounts of amino acids. In the healthy kidney, the glomeruli filter all amino acids out of the blood, and the renal tubules then reabsorb over 95% of the filtered amino acids back into the blood.

Dent's disease is a rare X-linked recessive inherited condition that affects the proximal renal tubules of the kidney. It is one cause of Fanconi syndrome, and is characterized by tubular proteinuria, excess calcium in the urine, formation of calcium kidney stones, nephrocalcinosis, and chronic kidney failure.

Sodium-dependent neutral amino acid transporter B(0)AT1 is a protein that in humans is encoded by the SLC6A19 gene.

Hypertryptophanemia is a rare autosomal recessive metabolic disorder that results in a massive buildup of the amino acid tryptophan in the blood, with associated symptoms and tryptophanuria.
Iminoglycinuria is an autosomal recessive disorder of renal tubular transport affecting reabsorption of the amino acid glycine, and the imino acids proline and hydroxyproline. This results in excess urinary excretion of all three acids.
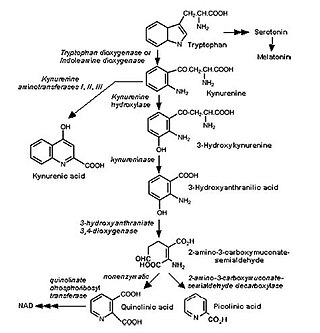
The kynurenine pathway is a metabolic pathway leading to the production of nicotinamide adenine dinucleotide (NAD+). Metabolites involved in the kynurenine pathway include tryptophan, kynurenine, kynurenic acid, xanthurenic acid, quinolinic acid, and 3-hydroxykynurenine. The kynurenine pathway is responsible for about 95% of total tryptophan catabolism. Disruption in the pathway is associated with certain genetic and psychiatric disorders.

Nutritional neuroscience is the scientific discipline that studies the effects various components of the diet such as minerals, vitamins, protein, carbohydrates, fats, dietary supplements, synthetic hormones, and food additives have on neurochemistry, neurobiology, behavior, and cognition.
Fanconi syndrome or Fanconi's syndrome is a syndrome of inadequate reabsorption in the proximal renal tubules of the kidney. The syndrome can be caused by various underlying congenital or acquired diseases, by toxicity, or by adverse drug reactions. It results in various small molecules of metabolism being passed into the urine instead of being reabsorbed from the tubular fluid. Fanconi syndrome affects the proximal tubules, namely, the proximal convoluted tubule (PCT), which is the first part of the tubule to process fluid after it is filtered through the glomerulus, and the proximal straight tubule, which leads to the descending limb of loop of Henle.
Dicarboxylic aminoaciduria is a rare form of aminoaciduria which is an autosomal recessive disorder of urinary glutamate and aspartate due to genetic errors related to transport of these amino acids. Mutations resulting in a lack of expression of the SLC1A1 gene, a member of the solute carrier family, are found to cause development of dicarboxylic aminoaciduria in humans. SLC1A1 encodes for EAAT3 which is found in the neurons, intestine, kidney, lung, and heart. EAAT3 is part of a family of high affinity glutamate transporters which transport both glutamate and aspartate across the plasma membrane.

Vitamin B3, colloquially referred to as niacin, is a vitamin family that includes three forms, or vitamers: niacin (nicotinic acid), nicotinamide (niacinamide), and nicotinamide riboside. All three forms of vitamin B3 are converted within the body to nicotinamide adenine dinucleotide (NAD). NAD is required for human life and people are unable to make it within their bodies without either vitamin B3 or tryptophan. Nicotinamide riboside was identified as a form of vitamin B3 in 2004.

