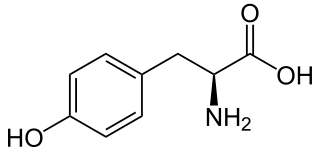
L-Tyrosine or tyrosine or 4-hydroxyphenylalanine is one of the 20 standard amino acids that are used by cells to synthesize proteins. It is a conditionally essential amino acid with a polar side group. The word "tyrosine" is from the Greek tyrós, meaning cheese, as it was first discovered in 1846 by German chemist Justus von Liebig in the protein casein from cheese. It is called tyrosyl when referred to as a functional group or side chain. While tyrosine is generally classified as a hydrophobic amino acid, it is more hydrophilic than phenylalanine. It is encoded by the codons UAC and UAU in messenger RNA.

Alkaptonuria is a rare inherited genetic disease which is caused by a mutation in the HGD gene for the enzyme homogentisate 1,2-dioxygenase ; if a person inherits an abnormal copy from both parents, the body accumulates an intermediate substance called homogentisic acid in the blood and tissues. Homogentisic acid and its oxidized form alkapton are excreted in the urine, giving it an unusually dark color. The accumulating homogentisic acid causes damage to cartilage and heart valves, as well as precipitating as kidney stones and stones in other organs. Symptoms usually develop in people over 30 years old, although the dark discoloration of the urine is present from birth.

Maple syrup urine disease (MSUD) is a rare, inherited metabolic disorder that affects the body's ability to metabolize amino acids due to a deficiency in the activity of the branched-chain alpha-ketoacid dehydrogenase (BCKAD) complex. It particularly affects the metabolism of amino acids—leucine, isoleucine, and valine. With MSUD, the body is not able to properly break down these amino acids, therefore leading to the amino acids to build up in urine and become toxic. The condition gets its name from the distinctive sweet odor of affected infants' urine and earwax due to the buildup of these amino acids.

Tyrosinemia or tyrosinaemia is an error of metabolism, usually inborn, in which the body cannot effectively break down the amino acid tyrosine. Symptoms of untreated tyrosinemia include liver and kidney disturbances. Without treatment, tyrosinemia leads to liver failure. Today, tyrosinemia is increasingly detected on newborn screening tests before any symptoms appear. With early and lifelong management involving a low-protein diet, special protein formula, and sometimes medication, people with tyrosinemia develop normally, are healthy, and live normal lives.

Glycine encephalopathy is a rare autosomal recessive disorder of glycine metabolism. After phenylketonuria, glycine encephalopathy is the second most common disorder of amino acid metabolism. The disease is caused by defects in the glycine cleavage system, an enzyme responsible for glycine catabolism. There are several forms of the disease, with varying severity of symptoms and time of onset. The symptoms are exclusively neurological in nature, and clinically this disorder is characterized by abnormally high levels of the amino acid glycine in bodily fluids and tissues, especially the cerebrospinal fluid.

4-Hydroxyphenylpyruvate dioxygenase (HPPD), also known as α-ketoisocaproate dioxygenase, is an Fe(II)-containing non-heme oxygenase that catalyzes the second reaction in the catabolism of tyrosine - the conversion of 4-hydroxyphenylpyruvate into homogentisate. HPPD also catalyzes the conversion of phenylpyruvate to 2-hydroxyphenylacetate and the conversion of α-ketoisocaproate to β-hydroxy β-methylbutyrate. HPPD is an enzyme that is found in nearly all aerobic forms of life.

Histidinemia is a rare autosomal recessive metabolic disorder caused by a deficiency of the enzyme histidase. Histidase is needed for the metabolism of the amino acid histidine. Although originally thought to be linked to multiple developmental disorders histidinemia is now accepted as a relatively benign disorder, leading to a reduction in the prevalence of neonatal screening procedures.

Nitisinone, sold under the brand name Orfadin among others, is a medication used to slow the effects of hereditary tyrosinemia type 1 (HT-1).
Methylcrotonyl CoA carboxylase is a biotin-requiring enzyme located in the mitochondria. MCC uses bicarbonate as a carboxyl group source to catalyze the carboxylation of a carbon adjacent to a carbonyl group performing the fourth step in processing leucine, an essential amino acid.
3-Hydroxy-3-methylglutaryl-CoA lyase is an enzyme (EC 4.1.3.4 that in human is encoded by the HMGCL gene located on chromosome 1. It is a key enzyme in ketogenesis. It is a ketogenic enzyme in the liver that catalyzes the formation of acetoacetate from HMG-CoA within the mitochondria. It also plays a prominent role in the catabolism of the amino acid leucine.

Homogentisate 1,2-dioxygenase (homogentisic acid oxidase, homogentisate oxidase, homogentisicase) is an enzyme which catalyzes the conversion of homogentisate to 4-maleylacetoacetate. Homogentisate 1,2-dioxygenase or HGD is involved in the catabolism of aromatic rings, more specifically in the breakdown of the amino acids tyrosine and phenylalanine. HGD appears in the metabolic pathway of tyrosine and phenylalanine degradation once the molecule homogentisate is produced. Homogentisate reacts with HGD to produce maleylacetoacetate, which then is further used in the metabolic pathway. HGD requires the use of Fe2+ and O2 in order to cleave the aromatic ring of homogentisate.

Hypermethioninemia is an excess of the amino acid methionine, in the blood. This condition can occur when methionine is not broken down properly in the body.

4-Hydroxyphenylpyruvic acid (4-HPPA) is an intermediate in the metabolism of the amino acid phenylalanine. The aromatic side chain of phenylalanine is hydroxylated by the enzyme phenylalanine hydroxylase to form tyrosine. The conversion from tyrosine to 4-HPPA is in turn catalyzed by tyrosine aminotransferase. Additionally, 4-HPPA can be converted to homogentisic acid which is one of the precursors to ochronotic pigment.

Tyrosine aminotransferase is an enzyme present in the liver and catalyzes the conversion of tyrosine to 4-hydroxyphenylpyruvate.

Fumarylacetoacetase is an enzyme that in humans is encoded by the FAH gene located on chromosome 15. The enzyme is involved in the catabolism of the amino acid tyrosine in humans.

Pyruvate dehydrogenase E1 component subunit alpha, somatic form, mitochondrial is an enzyme that in humans is encoded by the PDHA1 gene.The pyruvate dehydrogenase complex is a nuclear-encoded mitochondrial matrix multienzyme complex that provides the primary link between glycolysis and the tricarboxylic acid (TCA) cycle by catalyzing the irreversible conversion of pyruvate into acetyl-CoA. The PDH complex is composed of multiple copies of 3 enzymes: E1 (PDHA1); dihydrolipoyl transacetylase (DLAT) ; and dihydrolipoyl dehydrogenase (DLD). The E1 enzyme is a heterotetramer of 2 alpha and 2 beta subunits. The E1-alpha subunit contains the E1 active site and plays a key role in the function of the PDH complex.

In enzymology, maleylacetoacetate isomerase is an enzyme that catalyzes the chemical reaction
Tyrosinemia type III is a rare disorder caused by a deficiency of the enzyme 4-hydroxyphenylpyruvate dioxygenase, encoded by the gene HPD. This enzyme is abundant in the liver, and smaller amounts are found in the kidneys. It is one of a series of enzymes needed to break down tyrosine. Specifically, 4-hydroxyphenylpyruvate dioxygenase converts a tyrosine byproduct called 4-hydroxyphenylpyruvate to homogentisic acid. Characteristic features of type III tyrosinemia include mild mental retardation, seizures, and periodic loss of balance and coordination. Type III tyrosinemia is very rare; only a few cases have been reported.

Tyrosinemia type I is a genetic disorder that disrupts the metabolism of the amino acid tyrosine, resulting in damage primarily to the liver along with the kidneys and peripheral nerves. The inability of cells to process tyrosine can lead to chronic liver damage ending in liver failure, as well as renal disease and rickets. Symptoms such as poor growth and enlarged liver are associated with the clinical presentation of the disease. If not detected via newborn screening and management not begun before symptoms appear, clinical manifestation of disease occurs typically within the first two years of life. The severity of the disease is correlated with the timing of onset of symptoms, earlier being more severe. If diagnosed through newborn screening prior to clinical manifestation, and well managed with diet and medication, normal growth and development is possible.

Tyrosine hydroxylase deficiency (THD) is a disorder caused by disfunction of tyrosine hydroxylase, an enzyme involved in the biosynthesis of dopamine. This condition is one of the causes of dopa-responsive dystonia.
