Propionic acidemia, also known as propionic aciduria or propionyl-CoA carboxylase deficiency, is a rare autosomal recessive metabolic disorder, classified as a branched-chain organic acidemia.

Isovaleric acidemia is a rare autosomal recessive metabolic disorder which disrupts or prevents normal metabolism of the branched-chain amino acid leucine. It is a classical type of organic acidemia.

Maple syrup urine disease (MSUD) is a rare, inherited metabolic disorder that affects the body’s ability to metabolize amino acids due to a deficiency in the activity of the branched-chain alpha-ketoacid dehydrogenase (BCKAD) complex. It particularly affects the metabolism of amino acids- leucine, isoleucine, and valine. With MSUD, the body is not able to properly break down these amino acids, therefore leading to the amino acids to build up in urine and become toxic. The condition gets its name from the distinctive sweet odor of affected infants' urine and earwax due to the buildup of these amino acids.
In biochemistry and metabolism, beta oxidation (also β-oxidation) is the catabolic process by which fatty acid molecules are broken down in the cytosol in prokaryotes and in the mitochondria in eukaryotes to generate acetyl-CoA. Acetyl-CoA enters the citric acid cycle, generating NADH and FADH2, which are electron carriers used in the electron transport chain. It is named as such because the beta carbon of the fatty acid chain undergoes oxidation and is converted to a carbonyl group to start the cycle all over again. Beta-oxidation is primarily facilitated by the mitochondrial trifunctional protein, an enzyme complex associated with the inner mitochondrial membrane, although very long chain fatty acids are oxidized in peroxisomes.
Glutaric acidemia type 1 (GA1) is an inherited disorder in which the body is unable to completely break down the amino acids lysine, hydroxylysine and tryptophan. Excessive levels of their intermediate breakdown products can accumulate and cause damage to the brain, but particularly the basal ganglia, which are regions that help regulate movement. GA1 causes secondary carnitine deficiency, as glutaric acid, like other organic acids, is detoxified by carnitine. Mental retardation may occur.

Carnitine palmitoyltransferase I deficiency is a rare metabolic disorder that prevents the body from converting certain fats called long-chain fatty acids(LCFA) into energy, particularly during periods without food. It is caused by a mutation in CPT1A on chromosome 11.
Carnitine palmitoyltransferase II deficiency, sometimes shortened to CPT-II or CPT2, is an autosomal recessively inherited genetic metabolic disorder characterized by an enzymatic defect that prevents long-chain fatty acids from being transported into the mitochondria for utilization as an energy source. The disorder presents in one of three clinical forms: lethal neonatal, severe infantile hepatocardiomuscular and myopathic.

Carnitine-acylcarnitine translocase deficiency is a rare, autosomal recessive metabolic disorder that prevents the body from converting long-chain fatty acids into energy, particularly during periods without food. Carnitine, a natural substance acquired mostly through the diet, is used by cells to process fats and produce energy. People with this disorder have a faulty enzyme that prevents long-chain fatty acids from being transported into the innermost part of the mitochondria for processing.
Mitochondrial trifunctional protein deficiency is an autosomal recessive fatty acid oxidation disorder that prevents the body from converting certain fats to energy, particularly during periods without food. People with this disorder have inadequate levels of an enzyme that breaks down a certain group of fats called long-chain fatty acids.
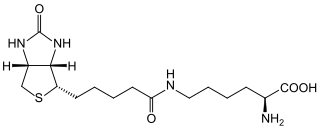
Biotinidase deficiency is an autosomal recessive metabolic disorder in which biotin is not released from proteins in the diet during digestion or from normal protein turnover in the cell. This situation results in biotin deficiency.
Glutaric acidemia type 2 is an autosomal recessive metabolic disorder that is characterised by defects in the ability of the body to use proteins and fats for energy. Incompletely processed proteins and fats can build up, leading to a dangerous chemical imbalance called acidosis. It is a metabolic myopathy, categorized under fatty acid metabolism disorder as that is the bioenergetic system that it affects the most. It also affects choline metabolism.
2-Methylbutyryl-CoA dehydrogenase deficiency is an autosomal recessive metabolic disorder. It causes the body to be unable to process the amino acid isoleucine properly. Initial case reports identified individuals with developmental delay and epilepsy, however most cases identified through newborn screening have been asymptomatic.
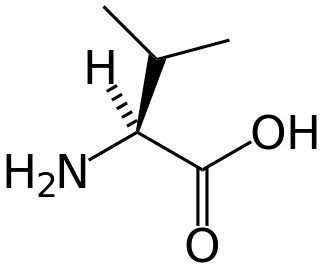
Isobutyryl-coenzyme A dehydrogenase deficiency is a rare metabolic disorder in which the body is unable to process certain amino acids properly.

Ornithine translocase deficiency, also called hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome, is a rare autosomal recessive urea cycle disorder affecting the enzyme ornithine translocase, which causes ammonia to accumulate in the blood, a condition called hyperammonemia.
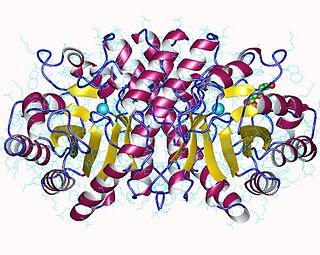
3-Hydroxy-3-methylglutaryl-CoA lyase is an enzyme (EC 4.1.3.4 that in human is encoded by the HMGCL gene located on chromosome 1. It is a key enzyme in ketogenesis. It is a ketogenic enzyme in the liver that catalyzes the formation of acetoacetate from HMG-CoA within the mitochondria. It also plays a prominent role in the catabolism of the amino acid leucine.

Hypermethioninemia is an excess of the amino acid methionine, in the blood. This condition can occur when methionine is not broken down properly in the body.
Hypervalinemia is a rare autosomal recessive metabolic disorder in which urinary and serum levels of the branched-chain amino acid valine are elevated, without related elevation of the branched-chain amino acids leucine and isoleucine. It is caused by a deficiency of the enzyme valine transaminase.
Organic acidemia is a term used to classify a group of metabolic disorders which disrupt normal amino acid metabolism, particularly branched-chain amino acids, causing a buildup of acids which are usually not present.
A broad classification for genetic disorders that result from an inability of the body to produce or utilize an enzyme or transport protein that is required to oxidize fatty acids. They are an inborn error of lipid metabolism, and when it affects the muscles also a metabolic myopathy.

Citrullinemia type I (CTLN1), also known as arginosuccinate synthetase deficiency, is a rare disease caused by a deficiency in argininosuccinate synthetase, an enzyme involved in excreting excess nitrogen from the body. There are mild and severe forms of the disease, which is one of the urea cycle disorders.

