
Methylmalonic acidemia, also called methylmalonic aciduria, is an autosomal recessive metabolic disorder that disrupts normal amino acid metabolism. It is a classical type of organic acidemia. The result of this condition is the inability to properly digest specific fats and proteins, which in turn leads to a buildup of a toxic level of methylmalonic acid in the blood.

Isovaleric acidemia is a rare autosomal recessive metabolic disorder which disrupts or prevents normal metabolism of the branched-chain amino acid leucine. It is a classical type of organic acidemia.

Maple syrup urine disease (MSUD) is an autosomal recessive metabolic disorder affecting branched-chain amino acids. It is one type of organic acidemia. The condition gets its name from the distinctive sweet odor of affected infants' urine and earwax, particularly prior to diagnosis and during times of acute illness.

Tyrosinemia or tyrosinaemia is an error of metabolism, usually inborn, in which the body cannot effectively break down the amino acid tyrosine. Symptoms of untreated tyrosinemia include liver and kidney disturbances. Without treatment, tyrosinemia leads to liver failure. Today, tyrosinemia is increasingly detected on newborn screening tests before any symptoms appear. With early and lifelong management involving a low-protein diet, special protein formula, and sometimes medication, people with tyrosinemia develop normally, are healthy, and live normal lives.

Systemic primary carnitine deficiency (SPCD) is an inborn error of fatty acid transport caused by a defect in the transporter responsible for moving carnitine across the plasma membrane. Carnitine is an important amino acid for fatty acid metabolism. When carnitine cannot be transported into tissues, fatty acid oxidation is impaired, leading to a variety of symptoms such as chronic muscle weakness, cardiomyopathy, hypoglycemia and liver dysfunction. The specific transporter involved with SPCD is OCTN2, coded for by the SLC22A5 gene located on chromosome 5. SPCD is inherited in an autosomal recessive manner, with mutated alleles coming from both parents.

Argininosuccinic aciduria is an inherited disorder that causes the accumulation of argininosuccinic acid in the blood and urine. Some patients may also have an elevation of ammonia, a toxic chemical, which can affect the nervous system. Argininosuccinic aciduria may become evident in the first few days of life because of high blood ammonia, or later in life presenting with "sparse" or "brittle" hair, developmental delay, and tremors.

Short-chain acyl-coenzyme A dehydrogenase deficiency (SCADD) is an autosomal recessive fatty acid oxidation disorder which affects enzymes required to break down a certain group of fats called short chain fatty acids.
2,4 Dienoyl-CoA reductase deficiency is an inborn error of metabolism resulting in defective fatty acid oxidation caused by a deficiency of the enzyme 2,4 Dienoyl-CoA reductase. Lysine degradation is also affected in this disorder leading to hyperlysinemia. The disorder is inherited in an autosomal recessive manner, meaning an individual must inherit mutations in NADK2, located at 5p13.2 from both of their parents. NADK2 encodes the mitochondrial NAD kinase. A defect in this enzyme leads to deficient mitochondrial nicotinamide adenine dinucleotide phosphate levels. 2,4 Dienoyl-CoA reductase, but also lysine degradation are performed by NADP-dependent oxidoreductases explaining how NADK2 deficiency can lead to multiple enzyme defects.

Glycine encephalopathy is a rare autosomal recessive disorder of glycine metabolism. After phenylketonuria, glycine encephalopathy is the second most common disorder of amino acid metabolism. The disease is caused by defects in the glycine cleavage system, an enzyme responsible for glycine catabolism. There are several forms of the disease, with varying severity of symptoms and time of onset. The symptoms are exclusively neurological in nature, and clinically this disorder is characterized by abnormally high levels of the amino acid glycine in bodily fluids and tissues, especially the cerebrospinal fluid.
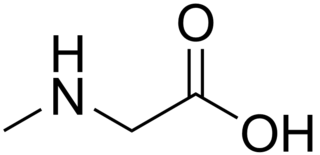
Sarcosinemia (SAR), also called hypersarcosinemia and SARDH deficiency, is a rare autosomal recessive metabolic disorder characterized by an increased concentration of sarcosine in blood plasma and urine ("sarcosinuria"). It can result from an inborn error of sarcosine metabolism, or from severe folate deficiency related to the folate requirement for the conversion of sarcosine to glycine. It is thought to be a relatively benign condition.

Urocanic acid is an intermediate in the catabolism of L-histidine.

Ornithine translocase deficiency, also called hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome, is a rare autosomal recessive urea cycle disorder affecting the enzyme ornithine translocase, which causes ammonia to accumulate in the blood, a condition called hyperammonemia.

Essential fructosuria, caused by a deficiency of the enzyme hepatic fructokinase, is a clinically benign condition characterized by the incomplete metabolism of fructose in the liver, leading to its excretion in urine. Fructokinase is the first enzyme involved in the degradation of fructose to fructose-1-phosphate in the liver.
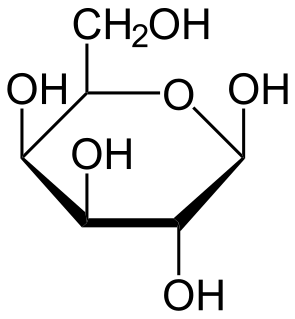
Galactose-1-phosphate uridylyltransferase deficiency(classic galactosemia) is the most common type of galactosemia, an inborn error of galactose metabolism, caused by a deficiency of the enzyme galactose-1-phosphate uridylyltransferase. It is an autosomal recessive metabolic disorder that can cause liver disease and death if untreated. Treatment of galactosemia is most successful if initiated early and includes dietary restriction of lactose intake. Because early intervention is key, galactosemia is included in newborn screening programs in many areas. On initial screening, which often involves measuring the concentration of galactose in blood, classic galactosemia may be indistinguishable from other inborn errors of galactose metabolism, including galactokinase deficiency and galactose epimerase deficiency. Further analysis of metabolites and enzyme activities are needed to identify the specific metabolic error.
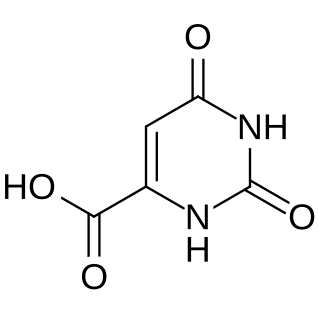
Orotic aciduria is a disease caused by an enzyme deficiency resulting in a decreased ability to synthesize pyrimidines. It was the first described enzyme deficiency of the de novo pyrimidine synthesis pathway.

Guanidinoacetate methyltransferase deficiency is an autosomal recessive cerebral creatine deficiency that primarily affects the nervous system and muscles. It is the first described disorder of creatine metabolism, and results from deficient activity of guanidinoacetate methyltransferase, an enzyme involved in the synthesis of creatine. Clinically, affected individuals often present with hypotonia, seizures and developmental delay. Diagnosis can be suspected on clinical findings, and confirmed by specific biochemical tests, brain magnetic resonance spectroscopy, or genetic testing. Biallelic pathogenic variants in GAMT are the underlying cause of the disorder. After GAMT deficiency is diagnosed, it can be treated by dietary adjustments, including supplementation with creatine. Treatment is highly effective if started early in life. If treatment is started late, it cannot reverse brain damage which has already taken place.

Hyperprolinemia is a condition which occurs when the amino acid proline is not broken down properly by the enzymes proline oxidase or pyrroline-5-carboxylate dehydrogenase, causing a buildup of proline in the body.
Urocanic aciduria is an autosomal recessive metabolic disorder caused by a deficiency of the enzyme urocanase. It is a secondary disorder of histidine metabolism.

Carnosinemia is a rare autosomal recessive metabolic disorder caused by a deficiency of carnosinase, a dipeptidase.
Ornithine aminotransferase deficiency is an inborn error of ornithine metabolism, caused by decreased activity of the enzyme ornithine aminotransferase. Biochemically, it can be detected by elevated levels of ornithine in the blood. Clinically, it presents initially with poor night vision, which slowly progresses to total blindness. It is believed to be inherited in an autosomal recessive manner. Approximately 200 known cases have been reported in the literature. The incidence is highest in Finland, estimated at 1:50,000.

