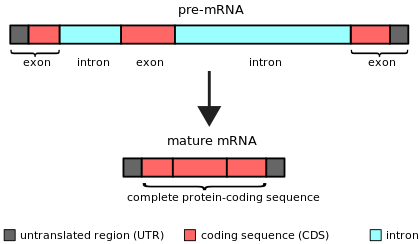
An intron is any nucleotide sequence within a gene that is not expressed or operative in the final RNA product. The word intron is derived from the term intragenic region, i.e., a region inside a gene.[1] The term intron refers to both the DNA sequence within a gene and the corresponding RNA sequence in RNA transcripts.[2] The non-intron sequences that become joined by this RNA processing to form the mature RNA are called exons.[3]
Introns are found in the genes of most eukaryotes and many eukaryotic viruses, and they can be located in both protein-coding genes and genes that function as RNA (noncoding genes). There are four main types of introns: tRNA introns, group I introns, group II introns, and spliceosomal introns (see below). Introns are rare in Bacteria and Archaea (prokaryotes).
Discovery and etymology
Introns were first discovered in protein-coding genes of adenovirus,[4][5] and were subsequently identified in genes encoding transfer RNA and ribosomal RNA genes. Introns are now known to occur within a wide variety of genes throughout organisms, bacteria,[6] and viruses within all of the biological kingdoms.
"The notion of the cistron [i.e., gene] ... must be replaced by that of a transcription unit containing regions which will be lost from the mature messenger – which I suggest we call introns (for intragenic regions) – alternating with regions which will be expressed – exons." (Gilbert 1978)
The term intron also refers to intracistron, i.e., an additional piece of DNA that arises within a cistron.[10]
Although introns are sometimes called intervening sequences,[11] the term "intervening sequence" can refer to any of several families of internal nucleic acid sequences that are not present in the final gene product, including inteins, untranslated regions (UTR), and nucleotides removed by RNA editing, in addition to introns.
Distribution
The frequency of introns within different genomes is observed to vary widely across the spectrum of biological organisms. For example, introns are extremely common within the nuclear genome of jawed vertebrates (e.g. humans, mice, and pufferfish (fugu)), where protein-coding genes almost always contain multiple introns, while introns are rare within the nuclear genes of some eukaryotic microorganisms,[12] for example baker's/brewer's yeast (Saccharomyces cerevisiae). In contrast, the mitochondrial genomes of vertebrates are entirely devoid of introns, while those of eukaryotic microorganisms may contain many introns.[13]
Simple illustration of an unspliced mRNA precursor, with two introns and three exons (top). After the introns have been removed via splicing, the mature mRNA sequence is ready for translation (bottom).
A particularly extreme case is the Drosophila DhDhc7 gene containing a ≥3.6 megabase (Mb) intron, which takes roughly three days to transcribe.[14][15] On the other extreme, a 2015 study suggests that the shortest known metazoan intron length is 30 base pairs (bp) belonging to the human MST1L gene.[16] The shortest known introns belong to the heterotrich ciliates, such as Stentor coeruleus, in which most (> 95%) introns are 15 or 16 bp long.[17]
Classification
Splicing of all intron-containing RNA molecules is superficially similar, as described above. However, different types of introns were identified through the examination of intron structure by DNA sequence analysis, together with genetic and biochemical analysis of RNA splicing reactions. At least four distinct classes of introns have been identified:
Self-splicing group II introns that are removed by RNA catalysis
Group III introns are proposed to be a fifth family, but little is known about the biochemical apparatus that mediates their splicing. They appear to be related to group II introns, and possibly to spliceosomal introns.[18]
Nuclear pre-mRNA introns (spliceosomal introns) are characterized by specific intron sequences located at the boundaries between introns and exons.[19] These sequences are recognized by spliceosomal RNA molecules when the splicing reactions are initiated.[20] In addition, they contain a branch point, a particular nucleotide sequence near the 3' end of the intron that becomes covalently linked to the 5' end of the intron during the splicing process, generating a branched (lariat)[clarification needed (complicated jargon)] intron. Apart from these three short conserved elements, nuclear pre-mRNA intron sequences are highly variable. Nuclear pre-mRNA introns are often much longer than their surrounding exons.
tRNA introns
Transfer RNA introns that depend upon proteins for removal occur at a specific location within the anticodon loop of unspliced tRNA precursors, and are removed by a tRNA splicing endonuclease. The exons are then linked together by a second protein, the tRNA splicing ligase.[21] Note that self-splicing introns are also sometimes found within tRNA genes.[22]
Group I and group II introns are found in genes encoding proteins (messenger RNA), transfer RNA and ribosomal RNA in a very wide range of living organisms.[23][24] Following transcription into RNA, group I and group II introns also make extensive internal interactions that allow them to fold into a specific, complex three-dimensional architecture. These complex architectures allow some group I and group II introns to be self-splicing, that is, the intron-containing RNA molecule can rearrange its own covalent structure so as to precisely remove the intron and link the exons together in the correct order. In some cases, particular intron-binding proteins are involved in splicing, acting in such a way that they assist the intron in folding into the three-dimensional structure that is necessary for self-splicing activity. Group I and group II introns are distinguished by different sets of internal conserved sequences and folded structures, and by the fact that splicing of RNA molecules containing group II introns generates branched introns (like those of spliceosomal RNAs), while group I introns use a non-encoded guanosine nucleotide (typically GTP) to initiate splicing, adding it on to the 5'-end of the excised intron.
On the accuracy of splicing
The spliceosome is a very complex structure containing up to one hundred proteins and five different RNAs. The substrate of the reaction is a long RNA molecule, and the transesterification reactions catalyzed by the spliceosome require the bringing together of sites that may be thousands of nucleotides apart.[25][26] All biochemical reactions are associated with known error rates – and the more complicated the reaction, the higher the error rate. Therefore, it is not surprising that the splicing reaction catalyzed by the spliceosome has a significant error rate even though there are spliceosome accessory factors that suppress the accidental cleavage of cryptic splice sites.[27]
Under ideal circumstances, the splicing reaction is likely to be 99.999% accurate (error rate of 10−5) and the correct exons will be joined and the correct intron will be deleted.[28] However, these ideal conditions require very close matches to the best splice site sequences and the absence of any competing cryptic splice site sequences within the introns, and those conditions are rarely met in large eukaryotic genes that may cover more than 40 kilobase pairs. Recent studies have shown that the actual error rate can be considerably higher than 10−5 and may be as high as 2% or 3% errors (error rate of 2 or 3×10−2) per gene.[29][30][31] Additional studies suggest that the error rate is no less than 0.1% per intron.[32][33] This relatively high level of splicing errors explains why most splice variants are rapidly degraded by nonsense-mediated decay.[34][35]
The presence of sloppy binding sites within genes causes splicing errors and it may seem strange that these sites haven't been eliminated by natural selection.[tone] The argument for their persistence is similar to the argument for junk DNA.[32][36]
Although mutations which create or disrupt binding sites may be slightly deleterious, the large number of possible such mutations makes it inevitable that some will reach fixation in a population. This is particularly relevant in species, such as humans, with relatively small long-term effective population sizes. It is plausible, then, that the human genome carries a substantial load of suboptimal sequences which cause the generation of aberrant transcript isoforms. In this study, we present direct evidence that this is indeed the case.[32]
While the catalytic reaction may be accurate enough for effective processing most of the time, the overall error rate may be partly limited by the fidelity of transcription because transcription errors will introduce mutations that create cryptic splice sites. In addition, the transcription error rate of 10−5 – 10−6 is high enough that one in every 25,000 transcribed exons will have an incorporation error in one of the splice sites leading to a skipped intron or a skipped exon. Almost all multi-exon genes will produce incorrectly spliced transcripts but the frequency of this background noise will depend on: the size of the genes, the number of introns, and the quality of the splice site sequences.[30][33]
In some cases, splice variants will be produced by mutations in the gene (DNA). These can be SNP polymorphisms that create a cryptic splice site or mutate a functional site. They can also be somatic cell mutations that affect splicing in a particular tissue or a cell line.[37][38][39] When the mutant allele is in a heterozygous state, this will result in production of two abundant splice variants: one functional, and one non-functional. In the homozygous state, the mutant alleles may cause a genetic disease, such as the hemophilia found in descendants of Queen Victoria, where a mutation in one of the introns in a blood clotting factor gene creates a cryptic 3' splice site, resulting in aberrant splicing.[40] A significant fraction of human deaths by disease may be caused by mutations that interfere with normal splicing, mostly by creating cryptic splice sites.[41][38]
Incorrectly spliced transcripts can easily be detected and their sequences entered into the online databases. They are usually described as "alternatively spliced" transcripts, which can be confusing because the term does not distinguish between real, biologically relevant, alternative splicing and processing noise due to splicing errors. One of the central issues in the field of alternative splicing is working out the differences between these two possibilities. Many scientists have argued that the null hypothesis should be splicing noise, putting the burden of proof on those who claim biologically relevant alternative splicing. According to those scientists, the claim of function must be accompanied by convincing evidence that multiple functional products are produced from the same gene.[42][43]
Biological functions and evolution
While introns do not encode protein products, they are integral to gene expression regulation. Some introns themselves encode functional RNAs through further processing after splicing to generate noncoding RNA molecules.[44]Alternative splicing is widely used to generate multiple proteins from a single gene. Furthermore, some introns play essential roles in a wide range of gene expression regulatory functions such as nonsense-mediated decay[45] and mRNA export.[46]
After the initial discovery of introns in protein-coding genes of the eukaryotic nucleus, there was significant debate as to whether introns in modern-day organisms were inherited from a common ancient ancestor (termed the introns-early hypothesis), or whether they appeared in genes rather recently in the evolutionary process (termed the introns-late hypothesis). Another theory is that the spliceosome and the intron-exon structure of genes is a relic of the RNA world (the introns-first hypothesis).[47] There is still considerable debate about the extent to which of these hypotheses is most correct but the popular consensus at the moment is that following the formation of the first eukaryotic cell group II introns from the bacterial endosymbiont invaded the host genome. In the beginning these self-splicing introns excised themselves from the mRNA precursor but over time some of them lost that ability and their excision had to be aided in trans by other group II introns. Eventually a number of specific trans-acting introns evolved and these became the precursors to the snRNAs of the spliceosome. The efficiency of splicing was improved by association with stabilizing proteins to form the primitive spliceosome.[48][49][50][51]
Early studies of genomic DNA sequences from a wide range of organisms show that the intron-exon structure of homologous genes in different organisms can vary widely.[52] More recent studies of entire eukaryotic genomes have now shown that the lengths and density (introns/gene) of introns varies considerably between related species. For example, while the human genome contains an average of 8.4 introns/gene (139,418 in the genome), the unicellular fungus Encephalitozoon cuniculi contains only 0.0075 introns/gene (15 introns in the genome).[53] Since eukaryotes arose from a common ancestor (common descent), there must have been extensive gain or loss of introns during evolutionary time.[54][55] This process is thought to be subject to selection, with a tendency towards intron gain in larger species due to their smaller population sizes, and the converse in smaller (particularly unicellular) species.[56] Biological factors also influence which genes in a genome lose or accumulate introns.[57][58][59]
Alternative splicing of exons within a gene after intron excision acts to introduce greater variability of protein sequences translated from a single gene, allowing multiple related proteins to be generated from a single gene and a single precursor mRNA transcript. The control of alternative RNA splicing is performed by a complex network of signaling molecules that respond to a wide range of intracellular and extracellular signals.
Introns contain several short sequences that are important for efficient splicing, such as acceptor and donor sites at either end of the intron as well as a branch point site, which are required for proper splicing by the spliceosome. Some introns are known to enhance the expression of the gene that they are contained in by a process known as intron-mediated enhancement (IME).
Actively transcribed regions of DNA frequently form R-loops that are vulnerable to DNA damage. In highly expressed yeast genes, introns inhibit R-loop formation and the occurrence of DNA damage.[60] Genome-wide analysis in both yeast and humans revealed that intron-containing genes have decreased R-loop levels and decreased DNA damage compared to intronless genes of similar expression.[60] Insertion of an intron within an R-loop prone gene can also suppress R-loop formation and recombination. Bonnet et al. (2017)[60] speculated that the function of introns in maintaining genetic stability may explain their evolutionary maintenance at certain locations, particularly in highly expressed genes.
Starvation adaptation
The physical presence of introns promotes cellular resistance to starvation via intron enhanced repression of ribosomal protein genes of nutrient-sensing pathways.[61]
As mobile genetic elements
Introns may be lost or gained over evolutionary time, as shown by many comparative studies of orthologous genes. Subsequent analyses have identified thousands of examples of intron loss and gain events, and it has been proposed that the emergence of eukaryotes, or the initial stages of eukaryotic evolution, involved an intron invasion.[62] Two definitive mechanisms of intron loss, reverse transcriptase-mediated intron loss (RTMIL) and genomic deletions, have been identified, and are known to occur.[63] The definitive mechanisms of intron gain, however, remain elusive and controversial. At least seven mechanisms of intron gain have been reported thus far: intron transposition, transposon insertion, tandem genomic duplication, intron transfer, intron gain during double-strand break repair (DSBR), insertion of a group II intron, and intronization. In theory it should be easiest to deduce the origin of recently gained introns due to the lack of host-induced mutations, yet even introns gained recently did not arise from any of the aforementioned mechanisms. These findings thus raise the question of whether or not the proposed mechanisms of intron gain fail to describe the mechanistic origin of many novel introns because they are not accurate mechanisms of intron gain, or if there are other, yet to be discovered, processes generating novel introns.[64]
In intron transposition, the most commonly purported intron gain mechanism, a spliced intron is thought to reverse splice into either its own mRNA or another mRNA at a previously intron-less position. This intron-containing mRNA is then reverse transcribed and the resulting intron-containing cDNA may then cause intron gain via complete or partial recombination with its original genomic locus.
Transposon insertions have been shown to generate thousands of new introns across diverse eukaryotic species.[65] Transposon insertions sometimes result in the duplication of this sequence on each side of the transposon. Such an insertion could intronize the transposon without disrupting the coding sequence when a transposon inserts into the sequence AGGT or encodes the splice sites within the transposon sequence. Where intron-generating transposons do not create target site duplications, elements include both splice sites GT (5') and AG (3') thereby splicing precisely without affecting the protein-coding sequence.[65] It is not yet understood why these elements are spliced, whether by chance, or by some preferential action by the transposon.
In tandem genomic duplication, due to the similarity between consensus donor and acceptor splice sites, which both closely resemble AGGT, the tandem genomic duplication of an exonic segment harboring an AGGT sequence generates two potential splice sites. When recognized by the spliceosome, the sequence between the original and duplicated AGGT will be spliced, resulting in the creation of an intron without alteration of the coding sequence of the gene. Double-stranded break repair via non-homologous end joining was recently identified as a source of intron gain when researchers identified short direct repeats flanking 43% of gained introns in Daphnia.[64] These numbers must be compared to the number of conserved introns flanked by repeats in other organisms, though, for statistical relevance. For group II intron insertion, the retrohoming of a group II intron into a nuclear gene was proposed to cause recent spliceosomal intron gain.
Intron transfer has been hypothesized to result in intron gain when a paralog or pseudogene gains an intron and then transfers this intron via recombination to an intron-absent location in its sister paralog. Intronization is the process by which mutations create novel introns from formerly exonic sequence. Thus, unlike other proposed mechanisms of intron gain, this mechanism does not require the insertion or generation of DNA to create a novel intron.[64]
The only hypothesized mechanism of recent intron gain lacking any direct evidence is that of group II intron insertion, which when demonstrated in vivo, abolishes gene expression.[66] Group II introns are therefore likely the presumed ancestors of spliceosomal introns, acting as site-specific retroelements, and are no longer responsible for intron gain.[67][68] Tandem genomic duplication is the only proposed mechanism with supporting in vivo experimental evidence: a short intragenic tandem duplication can insert a novel intron into a protein-coding gene, leaving the corresponding peptide sequence unchanged.[69] This mechanism also has extensive indirect evidence lending support to the idea that tandem genomic duplication is a prevalent mechanism for intron gain. The testing of other proposed mechanisms in vivo, particularly intron gain during DSBR, intron transfer, and intronization, is possible, although these mechanisms must be demonstrated in vivo to solidify them as actual mechanisms of intron gain. Further genomic analyses, especially when executed at the population level, may then quantify the relative contribution of each mechanism, possibly identifying species-specific biases that may shed light on varied rates of intron gain amongst different species.[64]
1 2 "The notion of the cistron [i.e., gene] ... must be replaced by that of a transcription unit containing regions which will be lost from the mature messenger – which I suggest we call introns (for intragenic regions) – alternating with regions which will be expressed – exons." (Gilbert 1978) Gilbert W (February 1978). "Why genes in pieces?". Nature. 271 (5645): 501. Bibcode:1978Natur.271..501G. doi:10.1038/271501a0. PMID622185. S2CID4216649.
↑ Belfort M, Pedersen-Lane J, West D, Ehrenman K, Maley G, Chu F, Maley F (June 1985). "Processing of the intron-containing thymidylate synthase (td) gene of phage T4 is at the RNA level". Cell. 41 (2): 375–382. doi:10.1016/s0092-8674(85)80010-6. PMID3986907. S2CID27127017.
↑ Copertino DW, Hallick RB (December 1993). "Group II and group III introns of twintrons: potential relationships with nuclear pre-mRNA introns". Trends in Biochemical Sciences. 18 (12): 467–471. doi:10.1016/0968-0004(93)90008-b. PMID8108859.
↑ Padgett RA, Grabowski PJ, Konarska MM, Seiler S, Sharp PA (1986). "Splicing of messenger RNA precursors". Annual Review of Biochemistry. 55: 1119–1150. doi:10.1146/annurev.bi.55.070186.005351. PMID2943217.
↑ Roy SW, Gilbert W (March 2006). "The evolution of spliceosomal introns: patterns, puzzles and progress". Nature Reviews. Genetics. 7 (3): 211–221. doi:10.1038/nrg1807. PMID16485020. S2CID33672491.
↑ Jeffares DC, Mourier T, Penny D (January 2006). "The biology of intron gain and loss". Trends in Genetics. 22 (1): 16–22. doi:10.1016/j.tig.2005.10.006. PMID16290250.
Bruce Alberts, Alexander Johnson, Julian Lewis, Martin Raff, Keith Roberts, and Peter Walter Molecular Biology of the Cell, 2007, ISBN978-0-8153-4105-5. Fourth edition is available online through the NCBI Bookshelf: link
Jeremy M Berg, John L Tymoczko, and Lubert Stryer, Biochemistry 5th edition, 2002, W H Freeman. Available online through the NCBI Bookshelf: link
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.