Related Research Articles

Phenylketonuria (PKU) is an inborn error of metabolism that results in decreased metabolism of the amino acid phenylalanine. Untreated PKU can lead to intellectual disability, seizures, behavioral problems, and mental disorders. It may also result in a musty smell and lighter skin. A baby born to a mother who has poorly treated PKU may have heart problems, a small head, and low birth weight.
Phenylalanine is an essential α-amino acid with the formula C
9H
11NO
2. It can be viewed as a benzyl group substituted for the methyl group of alanine, or a phenyl group in place of a terminal hydrogen of alanine. This essential amino acid is classified as neutral, and nonpolar because of the inert and hydrophobic nature of the benzyl side chain. The L-isomer is used to biochemically form proteins coded for by DNA. Phenylalanine is a precursor for tyrosine, the monoamine neurotransmitters dopamine, norepinephrine (noradrenaline), and epinephrine (adrenaline), and the biological pigment melanin. It is encoded by the messenger RNA codons UUU and UUC.
Inborn errors of metabolism form a large class of genetic diseases involving congenital disorders of enzyme activities. The majority are due to defects of single genes that code for enzymes that facilitate conversion of various substances (substrates) into others (products). In most of the disorders, problems arise due to accumulation of substances which are toxic or interfere with normal function, or due to the effects of reduced ability to synthesize essential compounds. Inborn errors of metabolism are often referred to as congenital metabolic diseases or inherited metabolic disorders. Another term used to describe these disorders is "enzymopathies". This term was created following the study of biodynamic enzymology, a science based on the study of the enzymes and their products. Finally, inborn errors of metabolism were studied for the first time by British physician Archibald Garrod (1857–1936), in 1908. He is known for work that prefigured the "one gene–one enzyme" hypothesis, based on his studies on the nature and inheritance of alkaptonuria. His seminal text, Inborn Errors of Metabolism, was published in 1923.

Maple syrup urine disease (MSUD) is a rare, inherited metabolic disorder that affects the body's ability to metabolize amino acids due to a deficiency in the activity of the branched-chain alpha-ketoacid dehydrogenase (BCKAD) complex. It particularly affects the metabolism of amino acids—leucine, isoleucine, and valine. With MSUD, the body is not able to properly break down these amino acids, therefore leading to the amino acids to build up in urine and become toxic. The condition gets its name from the distinctive sweet odor of affected infants' urine and earwax due to the buildup of these amino acids.

Cystinosis is a lysosomal storage disease characterized by the abnormal accumulation of cystine, the oxidized dimer of the amino acid cysteine. It is a genetic disorder that follows an autosomal recessive inheritance pattern. It is a rare autosomal recessive disorder resulting from accumulation of free cystine in lysosomes, eventually leading to intracellular crystal formation throughout the body. Cystinosis is the most common cause of Fanconi syndrome in the pediatric age group. Fanconi syndrome occurs when the function of cells in renal tubules is impaired, leading to abnormal amounts of carbohydrates and amino acids in the urine, excessive urination, and low blood levels of potassium and phosphates.

Beta-ketothiolase deficiency is a rare, autosomal recessive metabolic disorder in which the body cannot properly process the amino acid isoleucine or the products of lipid breakdown. Along with SCOT deficiency, it belongs to a group of disorders called ketone utilisation disorders.

Hartnup disease is an autosomal recessive metabolic disorder affecting the absorption of nonpolar amino acids. Niacin is a precursor to nicotinamide, a necessary component of NAD+.
Glutaric acidemia type 1 (GA1) is an inherited disorder in which the body is unable to completely break down the amino acids lysine, hydroxylysine and tryptophan. Excessive levels of their intermediate breakdown products can accumulate and cause damage to the brain, but particularly the basal ganglia, which are regions that help regulate movement. GA1 causes secondary carnitine deficiency, as glutaric acid, like other organic acids, is detoxified by carnitine. Mental retardation may occur.
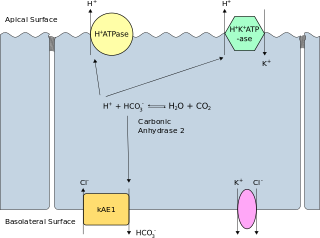
Band 3 anion transport protein, also known as anion exchanger 1 (AE1) or band 3 or solute carrier family 4 member 1 (SLC4A1), is a protein that is encoded by the SLC4A1 gene in humans.

Ornithine translocase deficiency, also called hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome, is a rare autosomal recessive urea cycle disorder affecting the enzyme ornithine translocase, which causes ammonia to accumulate in the blood, a condition called hyperammonemia.

Hypermethioninemia is an excess of the amino acid methionine, in the blood. This condition can occur when methionine is not broken down properly in the body.
Organic acidemia is a term used to classify a group of metabolic disorders which disrupt normal amino acid metabolism, particularly branched-chain amino acids, causing a buildup of acids which are usually not present.

Urocanic aciduria is an autosomal recessive metabolic disorder caused by a deficiency of the enzyme urocanase. It is a secondary disorder of histidine metabolism.
Inborn errors of renal tubular transport are metabolic disorders which lead to impairment in the ability of solutes, such as salts or amino acids, to be transported across the brush border of the renal tubule. This results in disruptions of renal reabsorption.
Hypertryptophanemia is a rare autosomal recessive metabolic disorder that results in a massive buildup of the amino acid tryptophan in the blood, with associated symptoms and tryptophanuria.
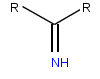
Iminoglycinuria is an autosomal recessive disorder of renal tubular transport affecting reabsorption of the amino acid glycine, and the imino acids proline and hydroxyproline. This results in excess urinary excretion of all three acids.
A low-sulfur diet is a diet with reduced sulfur content. Important dietary sources of sulfur and sulfur containing compounds may be classified as essential mineral, essential amino acid (methionine) and semi-essential amino acid.

Aminoacylase 1 deficiency is a rare inborn error of metabolism. To date only 21 cases have been described.
Dicarboxylic aminoaciduria is a rare form of aminoaciduria which is an autosomal recessive disorder of urinary glutamate and aspartate due to genetic errors related to transport of these amino acids. Mutations resulting in a lack of expression of the SLC1A1 gene, a member of the solute carrier family, are found to cause development of dicarboxylic aminoaciduria in humans. SLC1A1 encodes for EAAT3 which is found in the neurons, intestine, kidney, lung, and heart. EAAT3 is part of a family of high affinity glutamate transporters which transport both glutamate and aspartate across the plasma membrane.