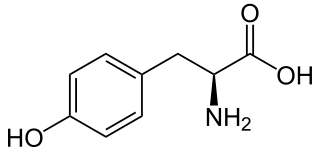
L-Tyrosine or tyrosine or 4-hydroxyphenylalanine is one of the 20 standard amino acids that are used by cells to synthesize proteins. It is a non-essential amino acid with a polar side group. The word "tyrosine" is from the Greek tyrós, meaning cheese, as it was first discovered in 1846 by German chemist Justus von Liebig in the protein casein from cheese. It is called tyrosyl when referred to as a functional group or side chain. While tyrosine is generally classified as a hydrophobic amino acid, it is more hydrophilic than phenylalanine. It is encoded by the codons UAC and UAU in messenger RNA.

Tyrosinemia or tyrosinaemia is an error of metabolism, usually inborn, in which the body cannot effectively break down the amino acid tyrosine. Symptoms of untreated tyrosinemia include liver and kidney disturbances. Without treatment, tyrosinemia leads to liver failure. Today, tyrosinemia is increasingly detected on newborn screening tests before any symptoms appear. With early and lifelong management involving a low-protein diet, special protein formula, and sometimes medication, people with tyrosinemia develop normally, are healthy, and live normal lives.

Homogentisic acid is a phenolic acid usually found in Arbutus unedo (strawberry-tree) honey. It is also present in the bacterial plant pathogen Xanthomonas campestris pv. phaseoli as well as in the yeast Yarrowia lipolytica where it is associated with the production of brown pigments. It is oxidatively dimerised to form hipposudoric acid, one of the main constituents of the 'blood sweat' of hippopotamuses.

4-Hydroxyphenylpyruvate dioxygenase (HPPD), also known as α-ketoisocaproate dioxygenase, is an Fe(II)-containing non-heme oxygenase that catalyzes the second reaction in the catabolism of tyrosine - the conversion of 4-hydroxyphenylpyruvate into homogentisate. HPPD also catalyzes the conversion of phenylpyruvate to 2-hydroxyphenylacetate and the conversion of α-ketoisocaproate to β-hydroxy β-methylbutyrate. HPPD is an enzyme that is found in nearly all aerobic forms of life.

Hawkinsinuria is an autosomal dominant metabolic disorder affecting the metabolism of tyrosine.

Nitisinone, sold under the brand name Orfadin among others, is a medication used to slow the effects of hereditary tyrosinemia type 1 (HT-1).

Homogentisate 1,2-dioxygenase (homogentisic acid oxidase, homogentisate oxidase, homogentisicase) is an enzyme which catalyzes the conversion of homogentisate to 4-maleylacetoacetate. Homogentisate 1,2-dioxygenase or HGD is involved in the catabolism of aromatic rings, more specifically in the breakdown of the amino acids tyrosine and phenylalanine. HGD appears in the metabolic pathway of tyrosine and phenylalanine degradation once the molecule homogentisate is produced. Homogentisate reacts with HGD to produce maleylacetoacetate, which then is further used in the metabolic pathway. HGD requires the use of Fe2+ and O2 in order to cleave the aromatic ring of homogentisate.

4-Hydroxyphenylpyruvic acid (4-HPPA) is an intermediate in the metabolism of the amino acid phenylalanine. The aromatic side chain of phenylalanine is hydroxylated by the enzyme phenylalanine hydroxylase to form tyrosine. The conversion from tyrosine to 4-HPPA is in turn catalyzed by tyrosine aminotransferase. Additionally, 4-HPPA can be converted to homogentisic acid which is one of the precursors to ochronotic pigment.

Tyrosine aminotransferase is an enzyme present in the liver and catalyzes the conversion of tyrosine to 4-hydroxyphenylpyruvate.

Fumarylacetoacetase is an enzyme that in humans is encoded by the FAH gene located on chromosome 15. The FAH gene is thought to be involved in the catabolism of the amino acid phenylalanine in humans.

Prephenate dehydrogenase is an enzyme found in the shikimate pathway, and helps catalyze the reaction from prephenate to tyrosine.
In enzymology, a 4-hydroxyphenylpyruvate oxidase (EC 1.2.3.13) is an enzyme that catalyzes the chemical reaction
In enzymology, a 4-hydroxymandelate synthase (EC 1.13.11.46) is an enzyme that catalyzes the chemical reaction

In enzymology, maleylacetoacetate isomerase is an enzyme that catalyzes the chemical reaction
Tyrosinemia type II is an autosomal recessive condition with onset between ages 2 and 4 years, when painful circumscribed calluses develop on the pressure points of the palm of the hand and sole of the foot.
4-Hydroxyphenylpyruvate dioxygenase (HPPD) inhibitors are a class of herbicides that prevent growth in plants by blocking 4-Hydroxyphenylpyruvate dioxygenase, an enzyme in plants that breaks down the amino acid tyrosine into molecules that are then used by plants to create other molecules that plants need. This process of breakdown, or catabolism, and making new molecules from the results, or biosynthesis, is something all living things do. HPPD inhibitors were first brought to market in 1980, although their mechanism of action was not understood until the late 1990s. They were originally used primarily in Japan in rice production, but since the late 1990s have been used in Europe and North America for corn, soybeans, and cereals, and since the 2000s have become more important as weeds have become resistant to glyphosate and other herbicides. Genetically modified crops are under development that include resistance to HPPD inhibitors. There is a pharmaceutical drug on the market, nitisinone, that was originally under development as an herbicide as a member of this class, and is used to treat an orphan disease, type I tyrosinemia.

Succinylacetone is a chemical compound that is formed by the oxidation of glycine and is a precursor of methylglyoxal. It is a pathognomonic compound found in the urine of patients with tyrosinemia type 1, which is due to congenital deficiency of an enzyme, fumarylacetoacetate hydrolase. This enzyme is involved in the catabolism of tyrosine, and if deficient, leads to accumulation of fumarylacetoacetate which is subsequently converted to succinylacetone which can be detected in the urine by GCMS. Succinylacetone also inhibits ALA dehydratase which increases ALA and precipitates acute neuropathic symptoms, similar to porphyria.

4-Hydroxyphenylglycine (HPG) is a non-proteogenic amino acid found in vancomycin and related glycopeptides. HPG is synthesized from the shikimic acid pathway and requires four enzymes to synthesize: Both L- and D-HPG are used in the vancomycin class of antibiotics. Tyrosine, a similar amino acid, differs by a methylene group (CH2) between the aromatic ring and the alpha carbon.

Tyrosinemia type I is a genetic disorder that disrupts the metabolism of the amino acid tyrosine, resulting in damage primarily to the liver along with the kidneys and peripheral nerves. The inability of cells to process tyrosine can lead to chronic liver damage ending in liver failure, as well as renal disease and rickets. Symptoms such as poor growth and enlarged liver are associated with the clinical presentation of the disease. If not detected via newborn screening and management not begun before symptoms appear, clinical manifestation of disease occurs typically within the first two years of life. The severity of the disease is correlated with the timing of onset of symptoms, earlier being more severe. If diagnosed through newborn screening prior to clinical manifestation, and well managed with diet and medication, normal growth and development is possible.

