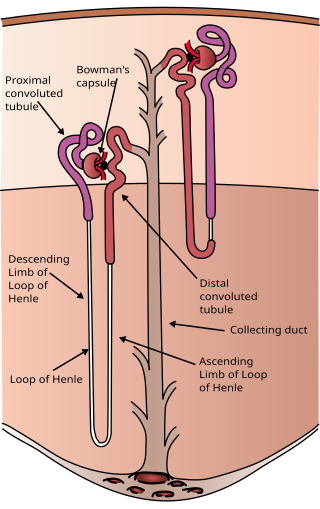
The nephron is the minute or microscopic structural and functional unit of the kidney. It is composed of a renal corpuscle and a renal tubule. The renal corpuscle consists of a tuft of capillaries called a glomerulus and a cup-shaped structure called Bowman's capsule. The renal tubule extends from the capsule. The capsule and tubule are connected and are composed of epithelial cells with a lumen. A healthy adult has 1 to 1.5 million nephrons in each kidney. Blood is filtered as it passes through three layers: the endothelial cells of the capillary wall, its basement membrane, and between the podocyte foot processes of the lining of the capsule. The tubule has adjacent peritubular capillaries that run between the descending and ascending portions of the tubule. As the fluid from the capsule flows down into the tubule, it is processed by the epithelial cells lining the tubule: water is reabsorbed and substances are exchanged ; first with the interstitial fluid outside the tubules, and then into the plasma in the adjacent peritubular capillaries through the endothelial cells lining that capillary. This process regulates the volume of body fluid as well as levels of many body substances. At the end of the tubule, the remaining fluid—urine—exits: it is composed of water, metabolic waste, and toxins.

The proximal tubule is the segment of the nephron in kidneys which begins from the renal pole of the Bowman's capsule to the beginning of loop of Henle. At this location, the glomerular parietal epithelial cells (PECs) lining bowman’s capsule abruptly transition to proximal tubule epithelial cells (PTECs). The proximal tubule can be further classified into the proximal convoluted tubule (PCT) and the proximal straight tubule (PST).

Hypertensive kidney disease is a medical condition referring to damage to the kidney due to chronic high blood pressure. It manifests as hypertensive nephrosclerosis. It should be distinguished from renovascular hypertension, which is a form of secondary hypertension, and thus has opposite direction of causation.
Acute tubular necrosis (ATN) is a medical condition involving the death of tubular epithelial cells that form the renal tubules of the kidneys. Because necrosis is often not present, the term acute tubular injury (ATI) is preferred by pathologists over the older name acute tubular necrosis (ATN). ATN presents with acute kidney injury (AKI) and is one of the most common causes of AKI. Common causes of ATN include low blood pressure and use of nephrotoxic drugs. The presence of "muddy brown casts" of epithelial cells found in the urine during urinalysis is pathognomonic for ATN. Management relies on aggressive treatment of the factors that precipitated ATN. Because the tubular cells continually replace themselves, the overall prognosis for ATN is quite good if the underlying cause is corrected, and recovery is likely within 7 to 21 days.

Cystinosis is a lysosomal storage disease characterized by the abnormal accumulation of cystine, the oxidized dimer of the amino acid cysteine. It is a genetic disorder that follows an autosomal recessive inheritance pattern. It is a rare autosomal recessive disorder resulting from accumulation of free cystine in lysosomes, eventually leading to intracellular crystal formation throughout the body. Cystinosis is the most common cause of Fanconi syndrome in the pediatric age group. Fanconi syndrome occurs when the function of cells in renal tubules is impaired, leading to abnormal amounts of carbohydrates and amino acids in the urine, excessive urination, and low blood levels of potassium and phosphates.
Hypoaldosteronism is an endocrinological disorder characterized by decreased levels of the hormone aldosterone. Similarly, isolated hypoaldosteronism is the condition of having lowered aldosterone without corresponding changes in cortisol.
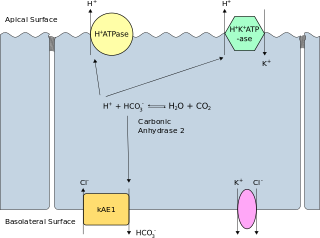
Band 3 anion transport protein, also known as anion exchanger 1 (AE1) or band 3 or solute carrier family 4 member 1 (SLC4A1), is a protein that is encoded by the SLC4A1 gene in humans.

Renal tubular acidosis (RTA) is a medical condition that involves an accumulation of acid in the body due to a failure of the kidneys to appropriately acidify the urine. In renal physiology, when blood is filtered by the kidney, the filtrate passes through the tubules of the nephron, allowing for exchange of salts, acid equivalents, and other solutes before it drains into the bladder as urine. The metabolic acidosis that results from RTA may be caused either by insufficient secretion of hydrogen ions into the latter portions of the nephron or by failure to reabsorb sufficient bicarbonate ions from the filtrate in the early portion of the nephron. Although a metabolic acidosis also occurs in those with chronic kidney disease, the term RTA is reserved for individuals with poor urinary acidification in otherwise well-functioning kidneys. Several different types of RTA exist, which all have different syndromes and different causes. RTA is usually an incidental finding based on routine blood draws that show abnormal results. Clinically, patients may present with vague symptoms such as dehydration, mental status changes, or delayed growth in adolescents.
Bartter syndrome (BS) is a rare inherited disease characterised by a defect in the thick ascending limb of the loop of Henle, which results in low potassium levels (hypokalemia), increased blood pH (alkalosis), and normal to low blood pressure. There are two types of Bartter syndrome: neonatal and classic. A closely associated disorder, Gitelman syndrome, is milder than both subtypes of Bartter syndrome.
A countercurrent mechanism system is a mechanism that expends energy to create a concentration gradient.

Oculocerebrorenal syndrome is a rare X-linked recessive disorder characterized by congenital cataracts, hypotonia, intellectual disability, proximal tubular acidosis, aminoaciduria and low-molecular-weight proteinuria. Lowe syndrome can be considered a cause of Fanconi syndrome.

Papillorenal syndrome is an autosomal dominant genetic disorder marked by underdevelopment (hypoplasia) of the kidney and colobomas of the optic nerve.
Dent's disease is a rare X-linked recessive inherited condition that affects the proximal renal tubules of the kidney. It is one cause of Fanconi syndrome, and is characterized by tubular proteinuria, excess calcium in the urine, formation of calcium kidney stones, nephrocalcinosis, and chronic kidney failure.

Medullary sponge kidney is a congenital disorder of the kidneys characterized by cystic dilatation of the collecting tubules in one or both kidneys. Individuals with medullary sponge kidney are at increased risk for kidney stones and urinary tract infection (UTI). Patients with MSK typically pass twice as many stones per year as do other stone formers without MSK. While having a low morbidity rate, as many as 10% of patients with MSK have an increased risk of morbidity associated with frequent stones and UTIs. While many patients report increased chronic kidney pain, the source of the pain, when a UTI or blockage is not present, is unclear at this time. Renal colic is present in 55% of patients. Women with MSK experience more stones, UTIs, and complications than men. MSK was previously believed not to be hereditary but there is more evidence coming forth that may indicate otherwise.
In renal physiology, renal sodium reabsorption refers to the process by which the kidneys, having filtered out waste products from the blood to be excreted as urine, re-absorb sodium ions (Na2+) from the waste. It uses Na-H antiport, Na-glucose symport, sodium ion channels (minor). It is stimulated by angiotensin II and aldosterone, and inhibited by atrial natriuretic peptide.
Iminoglycinuria is an autosomal recessive disorder of renal tubular transport affecting reabsorption of the amino acid glycine, and the imino acids proline and hydroxyproline. This results in excess urinary excretion of all three acids.
Fanconi syndrome or Fanconi's syndrome is a syndrome of inadequate reabsorption in the proximal renal tubules of the kidney. The syndrome can be caused by various underlying congenital or acquired diseases, by toxicity, or by adverse drug reactions. It results in various small molecules of metabolism being passed into the urine instead of being reabsorbed from the tubular fluid. Fanconi syndrome affects the proximal tubules, namely, the proximal convoluted tubule (PCT), which is the first part of the tubule to process fluid after it is filtered through the glomerulus, and the proximal straight tubule, which leads to the descending limb of loop of Henle.

Distal renal tubular acidosis (dRTA) is the classical form of RTA, being the first described. Distal RTA is characterized by a failure of acid secretion by the alpha intercalated cells of the distal tubule and cortical collecting duct of the distal nephron. This failure of acid secretion may be due to a number of causes. It leads to relatively alkaline urine, due to the kidney's inability to acidify the urine to a pH of less than 5.3.
Proximal renal tubular acidosis (pRTA) or type 2 renal tubular acidosis (RTA) is a type of RTA caused by a failure of the proximal tubular cells to reabsorb filtered bicarbonate from the urine, leading to urinary bicarbonate wasting and subsequent acidemia. The distal intercalated cells function normally, so the acidemia is less severe than dRTA and the urine can acidify to a pH of less than 5.3. pRTA also has several causes, and may occasionally be present as a solitary defect, but is usually associated with a more generalised dysfunction of the proximal tubular cells called Fanconi syndrome where there is also phosphaturia, glycosuria, aminoaciduria, uricosuria and tubular proteinuria.