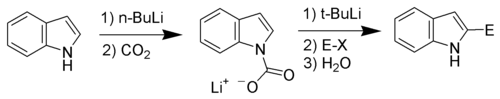
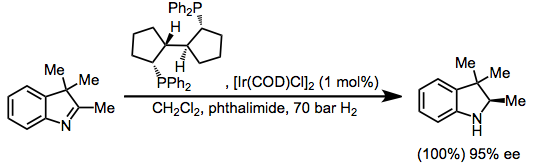
| 5-HT1 | | 5-HT1A | - Agonists: 4-F-5-MeO-pyr-T
- 5-MeO-pip-T
- 5-MeO-pyr-T
- 8-OH-DPAT
- Adatanserin
- Amphetamine
- Antidepressants (e.g., etoperidone, hydroxynefazodone, nefazodone, trazodone, triazoledione, vilazodone, vortioxetine)
- Atypical antipsychotics (e.g., aripiprazole, asenapine, brexpiprazole, cariprazine, clozapine, lurasidone, quetiapine, ziprasidone)
- Azapirones (e.g., buspirone, eptapirone, gepirone, perospirone, tandospirone)
- Bay R 1531
- Befiradol
- BMY-14802
- Cannabidiol
- Dimemebfe
- Dopamine
- Ebalzotan
- Eltoprazine
- Enciprazine
- Ergolines (e.g., bromocriptine, cabergoline, dihydroergotamine, ergotamine, lisuride, LSD, methylergometrine (methylergonovine), methysergide, pergolide)
- F-11461
- F-12826
- F-13714
- F-14679
- F-15063
- F-15599
- Flesinoxan
- Flibanserin
- Flumexadol
- Hypidone
- Lesopitron
- LY-293284
- LY-301317
- mCPP
- MKC-242
- Naluzotan
- NBUMP
- Osemozotan
- Oxaflozane
- Pardoprunox
- Piclozotan
- Rauwolscine
- Repinotan
- Roxindole
- RU-24969
- S-14506
- S-14671
- S-15535
- Sarizotan
- Serotonin (5-HT)
- SSR-181507
- Sunepitron
- Tryptamines (e.g., 5-CT, 5-MeO-DMT, 5-MT, bufotenin, DMT, indorenate, N-Me-5-HT, psilocin, psilocybin)
- TGBA01AD
- TMU4142
- U-92016-A
- Urapidil
- Vilazodone
- Xaliproden
- Yohimbine
| - Positive allosteric modulators: Oleamide
| - Antagonists: Atypical antipsychotics (e.g., iloperidone, risperidone, sertindole)
- AV965
- Beta blockers (e.g., alprenolol, carteolol, cyanopindolol, iodocyanopindolol, isamoltane, oxprenolol, penbutolol, pindobind, pindolol, propranolol, tertatolol)
- BMY-7378
- CSP-2503
- Dotarizine
- Ergolines (e.g., metergoline)
- FCE-24379
- Flopropione
- GR-46611
- Isamoltane
- Lecozotan
- Mefway
- Metitepine (methiothepin)
- MIN-117 (WF-516)
- MPPF
- NAN-190
- Robalzotan
- S-15535
- SB-649915
- SDZ 216-525
- Spiperone
- Spiramide
- Spiroxatrine
- UH-301
- WAY-100135
- WAY-100635
- Xylamidine
| |
|
|---|
| 5-HT1B | - Agonists: Anpirtoline
- AZ10419369
- Benzofurans (e.g., 5-MAPB, 6-MAPB, BK-5-MAPB, BK-6-MAPB)
- Benzothiophenes (e.g., 5-MAPBT, 6-MAPBT, BK-5-MAPBT)
- CGS-12066A
- CP-93129
- CP-94253
- CP-122288
- CP-135807
- Eltoprazine
- Ergolines (e.g., bromocriptine, dihydroergotamine, ergotamine, methylergometrine (methylergonovine), methysergide, pergolide)
- mCPP
- Methylenedioxyphenethylamines (e.g., MDMA, methylone)
- PZKKN-94
- RU-24969
- Serotonin (5-HT)
- Triptans (e.g., avitriptan, donitriptan, eletriptan, sumatriptan, zolmitriptan)
- TFMPP
- Tryptamines (e.g., 5-BT, 5-CT, 5-MT, DMT)
- Vortioxetine
| | |
|
|---|
| 5-HT1D | - Agonists: CP-122288
- CP-135807
- CP-286601
- Ergolines (e.g., bromocriptine, cabergoline, dihydroergotamine, ergotamine, LSD, methysergide)
- GR-46611
- L-694247
- L-772405
- mCPP
- PNU-109291
- PNU-142633
- Serotonin (5-HT)
- TGBA01AD
- Triptans (e.g., almotriptan, avitriptan, donitriptan, eletriptan, frovatriptan, naratriptan, rizatriptan, sumatriptan, zolmitriptan)
- Tryptamines (e.g., 5-BT, 5-CT, 5-Et-DMT, 5-MT, 5-(nonyloxy)tryptamine, DMT)
| | |
|
|---|
| 5-HT1E | |
|---|
| 5-HT1F | |
|---|
|
|---|
| 5-HT2 | | 5-HT2A | - Agonists: 25H/NB series (e.g., 25I-NBF, 25I-NBMD, 25I-NBOH, 25I-NBOMe, 25B-NBOMe, 25C-NBOMe, 25TFM-NBOMe, 2CBCB-NBOMe, 25CN-NBOH, 2CBFly-NBOMe)
- 2Cs (e.g., 2C-B, 2C-E, 2C-I, 2C-T-2, 2C-T-7, 2C-T-21)
- 2C-B-FLY
- 2CB-Ind
- 5-Methoxytryptamines (5-MeO-DET, 5-MeO-DiPT, 5-MeO-DMT, 5-MeO-DPT, 5-MT)
- α-Alkyltryptamines (e.g., 5-Cl-αMT, 5-Fl-αMT, 5-MeO-αET, 5-MeO-αMT, α-Me-5-HT, αET, αMT)
- AL-34662
- AL-37350A
- Bromo-DragonFLY
- Dimemebfe
- DMBMPP
- DOx (e.g., DOB, DOC, DOI, DOM)
- Efavirenz
- Ergolines (e.g., 1P-LSD, ALD-52, bromocriptine, cabergoline, ergine (LSA), ergometrine (ergonovine), ergotamine, lisuride, LA-SS-Az, LSB, LSD, LSD-Pip, LSH, LSP, methylergometrine (methylergonovine), pergolide)
- Flumexadol
- IHCH-7113
- Jimscaline
- Lorcaserin
- MDxx (e.g., MDA (tenamfetamine), MDMA (midomafetamine), MDOH, MMDA)
- O-4310
- Oxaflozane
- PHA-57378
- PNU-22394
- PNU-181731
- RH-34
- SCHEMBL5334361
- Phenethylamines (e.g., lophophine, mescaline)
- Piperazines (e.g., BZP, quipazine, TFMPP)
- Serotonin (5-HT)
- TCB-2
- TFMFly
- Tryptamines (e.g., 5-BT, 5-CT, bufotenin, DET, DiPT, DMT, DPT, psilocin, psilocybin, tryptamine)
| | - Antagonists: 5-I-R91150
- 5-MeO-NBpBrT
- AC-90179
- Adatanserin
- Altanserin
- Antihistamines (e.g., cyproheptadine, hydroxyzine, ketotifen, perlapine)
- AMDA
- Atypical antipsychotics (e.g., amperozide, aripiprazole, asenapine, blonanserin, brexpiprazole, carpipramine, clocapramine, clorotepine, clozapine, fluperlapine, gevotroline, iloperidone, lurasidone, melperone, mosapramine, ocaperidone, olanzapine, paliperidone, quetiapine, risperidone, sertindole, zicronapine, ziprasidone, zotepine)
- Chlorprothixene
- Cinanserin
- CSP-2503
- Deramciclane
- Dotarizine
- Eplivanserin
- Ergolines (e.g., amesergide, LY-53857, LY-215840, mesulergine, metergoline, methysergide, sergolexole)
- Fananserin
- Flibanserin
- Glemanserin
- Irindalone
- Ketanserin
- KML-010
- Landipirdine
- LY-393558
- mCPP
- Medifoxamine
- Metitepine (methiothepin)
- MIN-117 (WF-516)
- Naftidrofuryl
- Nantenine
- Nelotanserin
- Opiranserin (VVZ-149)
- Pelanserin
- Phenoxybenzamine
- Pimavanserin
- Pirenperone
- Pizotifen
- Pruvanserin
- Rauwolscine
- Ritanserin
- Roluperidone
- S-14671
- Sarpogrelate
- Serotonin antagonists and reuptake inhibitors (e.g., etoperidone, hydroxynefazodone, lubazodone, mepiprazole, nefazodone, triazoledione, trazodone)
- SR-46349B
- TGBA01AD
- Teniloxazine
- Temanogrel
- Tetracyclic antidepressants (e.g., amoxapine, aptazapine, esmirtazapine, maprotiline, mianserin, mirtazapine)
- Tricyclic antidepressants (e.g., amitriptyline)
- Typical antipsychotics (e.g., chlorpromazine, fluphenazine, haloperidol, loxapine, perphenazine, pimozide, pipamperone, prochlorperazine, setoperone, spiperone, spiramide, thioridazine, thiothixene, trifluoperazine)
- Volinanserin
- Xylamidine
- Yohimbine
| |
|
|---|
| 5-HT2B | - Agonists: 4-Methylaminorex
- Aminorex
- Amphetamines (e.g., chlorphentermine, cloforex, dexfenfluramine, fenfluramine, levofenfluramine, norfenfluramine)
- BW-723C86
- DOx (e.g., DOB, DOC, DOI, DOM)
- Ergolines (e.g., cabergoline, dihydroergocryptine, dihydroergotamine, ergotamine, methylergometrine (methylergonovine), methysergide, pergolide)
- Lorcaserin
- MDxx (e.g., MDA (tenamfetamine), MDMA (midomafetamine), MDOH, MMDA)
- Piperazines (e.g., TFMPP)
- PNU-22394
- Ro60-0175
- Serotonin (5-HT)
- Tryptamines (e.g., 5-BT, 5-CT, 5-MT, α-Me-5-HT, bufotenin, DET, DiPT, DMT, DPT, psilocin, psilocybin, tryptamine)
| - Antagonists: Agomelatine
- Atypical antipsychotics (e.g., amisulpride, aripiprazole, asenapine, brexpiprazole, cariprazine, clozapine, N-desalkylquetiapine (norquetiapine), N-desmethylclozapine (norclozapine), olanzapine, pipamperone, quetiapine, risperidone, ziprasidone)
- Cyproheptadine
- EGIS-7625
- Ergolines (e.g., amesergide, bromocriptine, lisuride, LY-53857, LY-272015, mesulergine)
- Ketanserin
- LY-393558
- mCPP
- Metadoxine
- Metitepine (methiothepin)
- Pirenperone
- Pizotifen
- Propranolol
- PRX-08066
- Rauwolscine
- Ritanserin
- RS-127445
- Sarpogrelate
- SB-200646
- SB-204741
- SB-206553
- SB-215505
- SB-221284
- SB-228357
- SDZ SER-082
- Tegaserod
- Tetracyclic antidepressants (e.g., amoxapine, mianserin, mirtazapine)
- Trazodone
- Typical antipsychotics (e.g., chlorpromazine)
- TIK-301
- Yohimbine
| |
|
|---|
| 5-HT2C | - Agonists: 2Cs (e.g., 2C-B, 2C-E, 2C-I, 2C-T-2, 2C-T-7, 2C-T-21)
- 5-Methoxytryptamines (5-MeO-DET, 5-MeO-DiPT, 5-MeO-DMT, 5-MeO-DPT, 5-MT)
- α-Alkyltryptamines (e.g., 5-Cl-αMT, 5-Fl-αMT, 5-MeO-αET, 5-MeO-αMT, α-Me-5-HT, αET, αMT)
- A-372159
- AL-38022A
- Alstonine
- Centhaquine
- CP-809101
- Dimemebfe
- DOx (e.g., DOB, DOC, DOI, DOM)
- Ergolines (e.g., ALD-52, cabergoline, dihydroergotamine, ergine (LSA), ergotamine, lisuride, LA-SS-Az, LSB, LSD, LSD-Pip, LSH, LSP, pergolide)
- Flumexadol
- Lorcaserin
- MDxx (e.g., MDA (tenamfetamine), MDMA (midomafetamine), MDOH, MMDA)
- MK-212
- ORG-12962
- ORG-37684
- Oxaflozane
- PHA-57378
- Phenethylamines (e.g., lophophine, mescaline)
- Piperazines (e.g., aripiprazole, BZP, mCPP, quipazine, TFMPP)
- PNU-22394
- PNU-181731
- Ro60-0175
- Ro60-0213
- Serotonin (5-HT)
- Tryptamines (e.g., 5-BT, 5-CT, bufotenin, DET, DiPT, DMT, DPT, psilocin, psilocybin, tryptamine)
- Vabicaserin
- WAY-629
- WAY-161503
- YM-348
| | - Antagonists: Adatanserin
- Agomelatine
- Atypical antipsychotics (e.g., asenapine, clorotepine, clozapine, fluperlapine, iloperidone, melperone, olanzapine, paliperidone, quetiapine, risperidone, sertindole, ziprasidone, zotepine)
- Captodiame
- CEPC
- Cinanserin
- Cyproheptadine
- Deramciclane
- Desmetramadol
- Dotarizine
- Eltoprazine
- Ergolines (e.g., amesergide, bromocriptine, LY-53857, LY-215840, mesulergine, metergoline, methysergide, sergolexole)
- Etoperidone
- Fluoxetine
- FR-260010
- Irindalone
- Ketanserin
- Ketotifen
- Latrepirdine (dimebolin)
- Medifoxamine
- Metitepine (methiothepin)
- Nefazodone
- Pirenperone
- Pizotifen
- Propranolol
- Ritanserin
- RS-102221
- S-14671
- SB-200646
- SB-206553
- SB-221284
- SB-228357
- SB-242084
- SB-243213
- SDZ SER-082
- Tedatioxetine
- Tetracyclic antidepressants (e.g., amoxapine, aptazapine, esmirtazapine, maprotiline, mianserin, mirtazapine)
- TIK-301
- Tramadol
- Trazodone
- Tricyclic antidepressants (e.g., amitriptyline, nortriptyline)
- Typical antipsychotics (e.g., chlorpromazine, loxapine, pimozide, pipamperone, thioridazine)
- Xylamidine
| |
|
|---|
|
|---|
| 5-HT3 –7 | | 5-HT3 | - Agonists: Alcohols (e.g., butanol, ethanol (alcohol), trichloroethanol)
- m-CPBG
- Phenylbiguanide
- Piperazines (e.g., BZP, mCPP, quipazine)
- RS-56812
- Serotonin (5-HT)
- SR-57227
- SR-57227A
- Tryptamines (e.g., 2-Me-5-HT, 5-CT, bufotenidine (5-HTQ))
- Volatiles/gases (e.g., halothane, isoflurane, toluene, trichloroethane)
- YM-31636
| | - Antagonists: Alosetron
- Anpirtoline
- Arazasetron
- AS-8112
- Atypical antipsychotics (e.g., clozapine, olanzapine, quetiapine)
- Azasetron
- Batanopride
- Bemesetron (MDL-72222)
- Cilansetron
- CSP-2503
- Dazopride
- Dolasetron
- Galanolactone
- Granisetron
- Lerisetron
- Memantine
- Ondansetron
- Palonosetron
- Ramosetron
- Renzapride
- Ricasetron
- Tedatioxetine
- Tetracyclic antidepressants (e.g., amoxapine, mianserin, mirtazapine)
- Thujone
- Tropanserin
- Tropisetron
- Typical antipsychotics (e.g., loxapine)
- Volatiles/gases (e.g., nitrous oxide, sevoflurane, xenon)
- Vortioxetine
- Zacopride
- Zatosetron
| | |
|
|---|
| 5-HT4 | |
|---|
| 5-HT5A | |
|---|
| 5-HT6 | - Agonists: Ergolines (e.g., dihydroergocryptine, dihydroergotamine, ergotamine, lisuride, LSD, mesulergine, metergoline, methysergide)
- Hypidone
- Serotonin (5-HT)
- Tryptamines (e.g., 2-Me-5-HT, 5-BT, 5-CT, 5-MT, Bufotenin, E-6801, E-6837, EMD-386088, EMDT, LY-586713, N-Me-5-HT, ST-1936, tryptamine)
- WAY-181187
- WAY-208466
| - Antagonists: ABT-354
- Atypical antipsychotics (e.g., aripiprazole, asenapine, clorotepine, clozapine, fluperlapine, iloperidone, olanzapine, tiospirone)
- AVN-101
- AVN-211
- AVN-322
- AVN-397
- BGC20-760
- BVT-5182
- BVT-74316
- Cerlapirdine
- EGIS-12233
- GW-742457
- Idalopirdine
- Ketanserin
- Landipirdine
- Latrepirdine (dimebolin)
- Masupirdine
- Metitepine (methiothepin)
- MS-245
- PRX-07034
- PZKKN-94
- Ritanserin
- Ro 04-6790
- Ro 63-0563
- SB-258585
- SB-271046
- SB-357134
- SB-399885
- SB-742457
- Tetracyclic antidepressants (e.g., amoxapine, mianserin)
- Tricyclic antidepressants (e.g., amitriptyline, clomipramine, doxepin, nortriptyline)
- Typical antipsychotics (e.g., chlorpromazine, loxapine)
| |
|
|---|
| 5-HT7 | | - Antagonists: Atypical antipsychotics (e.g., amisulpride, aripiprazole, asenapine, brexpiprazole, clorotepine, clozapine, fluperlapine, olanzapine, risperidone, sertindole, tiospirone, ziprasidone, zotepine)
- Butaclamol
- DR-4485
- EGIS-12233
- Ergolines (e.g., 2-Br-LSD (BOL-148), amesergide, bromocriptine, cabergoline, dihydroergotamine, ergotamine, LY-53857, LY-215840, mesulergine, metergoline, methysergide, sergolexole)
- JNJ-18038683
- Ketanserin
- LY-215840
- Metitepine (methiothepin)
- Ritanserin
- SB-258719
- SB-258741
- SB-269970
- SB-656104
- SB-656104A
- SB-691673
- SLV-313
- SLV-314
- Spiperone
- SSR-181507
- Tetracyclic antidepressants (e.g., amoxapine, maprotiline, mianserin, mirtazapine)
- Tricyclic antidepressants (e.g., amitriptyline, clomipramine, imipramine)
- Typical antipsychotics (e.g., acetophenazine, chlorpromazine, chlorprothixene, fluphenazine, loxapine, pimozide)
- Vortioxetine
| - Negative allosteric modulators: Oleamide
| |
|
|---|
|
|---|