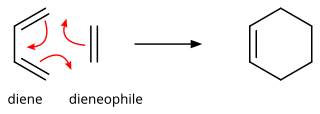
In organic chemistry, the Diels–Alder reaction is a chemical reaction between a conjugated diene and a substituted alkene, commonly termed the dienophile, to form a substituted cyclohexene derivative. It is the prototypical example of a pericyclic reaction with a concerted mechanism. More specifically, it is classified as a thermally-allowed [4+2] cycloaddition with Woodward–Hoffmann symbol [π4s + π2s]. It was first described by Otto Diels and Kurt Alder in 1928. For the discovery of this reaction, they were awarded the Nobel Prize in Chemistry in 1950. Through the simultaneous construction of two new carbon–carbon bonds, the Diels–Alder reaction provides a reliable way to form six-membered rings with good control over the regio- and stereochemical outcomes. Consequently, it has served as a powerful and widely applied tool for the introduction of chemical complexity in the synthesis of natural products and new materials. The underlying concept has also been applied to π-systems involving heteroatoms, such as carbonyls and imines, which furnish the corresponding heterocycles; this variant is known as the hetero-Diels–Alder reaction. The reaction has also been generalized to other ring sizes, although none of these generalizations have matched the formation of six-membered rings in terms of scope or versatility. Because of the negative values of ΔH° and ΔS° for a typical Diels–Alder reaction, the microscopic reverse of a Diels–Alder reaction becomes favorable at high temperatures, although this is of synthetic importance for only a limited range of Diels-Alder adducts, generally with some special structural features; this reverse reaction is known as the retro-Diels–Alder reaction.
Pyrrole is a heterocyclic, aromatic, organic compound, a five-membered ring with the formula C4H4NH. It is a colorless volatile liquid that darkens readily upon exposure to air. Substituted derivatives are also called pyrroles, e.g., N-methylpyrrole, C4H4NCH3. Porphobilinogen, a trisubstituted pyrrole, is the biosynthetic precursor to many natural products such as heme.

In organic chemistry, the Michael reaction or Michael 1,4 addition is a reaction between a Michael donor and a Michael acceptor to produce a Michael adduct by creating a carbon-carbon bond at the acceptor's β-carbon. It belongs to the larger class of conjugate additions and is widely used for the mild formation of carbon-carbon bonds.
The Wittig reaction or Wittig olefination is a chemical reaction of an aldehyde or ketone with a triphenyl phosphonium ylide called a Wittig reagent. Wittig reactions are most commonly used to convert aldehydes and ketones to alkenes. Most often, the Wittig reaction is used to introduce a methylene group using methylenetriphenylphosphorane (Ph3P=CH2). Using this reagent, even a sterically hindered ketone such as camphor can be converted to its methylene derivative.

The Michaelis–Arbuzov reaction is the chemical reaction of a trivalent phosphorus ester with an alkyl halide to form a pentavalent phosphorus species and another alkyl halide. The picture below shows the most common types of substrates undergoing the Arbuzov reaction; phosphite esters (1) react to form phosphonates (2), phosphonites (3) react to form phosphinates (4) and phosphinites (5) react to form phosphine oxides (6).
The Japp–Klingemann reaction is a chemical reaction used to synthesize hydrazones from β-keto-acids and aryl diazonium salts. The reaction is named after the chemists Francis Robert Japp and Felix Klingemann.
The Nenitzescu indole synthesis is a chemical reaction that forms 5-hydroxyindole derivatives from benzoquinone and β-aminocrotonic esters.

The Curtius rearrangement, first defined by Theodor Curtius in 1885, is the thermal decomposition of an acyl azide to an isocyanate with loss of nitrogen gas. The isocyanate then undergoes attack by a variety of nucleophiles such as water, alcohols and amines, to yield a primary amine, carbamate or urea derivative respectively. Several reviews have been published.

The Knorr pyrrole synthesis is a widely used chemical reaction that synthesizes substituted pyrroles (3). The method involves the reaction of an α-amino-ketone (1) and a compound containing an electron-withdrawing group α to a carbonyl group (2).
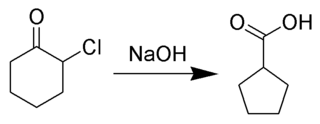
The Favorskii rearrangement is principally a rearrangement of cyclopropanones and α-halo ketones that leads to carboxylic acid derivatives. In the case of cyclic α-halo ketones, the Favorskii rearrangement constitutes a ring contraction. This rearrangement takes place in the presence of a base, sometimes hydroxide, to yield a carboxylic acid but most of the time either an alkoxide base or an amine to yield an ester or an amide, respectively. α,α'-Dihaloketones eliminate HX under the reaction conditions to give α,β-unsaturated carbonyl compounds.
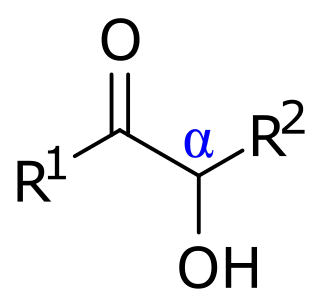
In organic chemistry, acyloins or α-hydroxy ketones are a class of organic compounds of the general form R−C(=O)−CR'(OH)−R", composed of a hydroxy group adjacent to a ketone group. The name acyloin is derived from the fact that they are formally derived from reductive coupling of carboxylic acyl groups. They are one of the two main classes of hydroxy ketones, distinguished by the position of the hydroxy group relative to the ketone; in this form, the hydroxy is on the alpha carbon, explaining the secondary name of α-hydroxy ketone.

The Darzens reaction is the chemical reaction of a ketone or aldehyde with an α-haloester in the presence of a base to form an α,β-epoxy ester, also called a "glycidic ester". This reaction was discovered by the organic chemist Auguste Georges Darzens in 1904.

The Hell–Volhard–Zelinsky halogenation reaction is a chemical transformation that involves the halogenation of a carboxylic acid at the α carbon. For this reaction to occur the α carbon must bear at least one proton. The reaction is named after the German chemists Carl Magnus von Hell (1849–1926) and Jacob Volhard (1834–1910) and the Russian chemist Nikolay Zelinsky (1861–1953).

The Wolff rearrangement is a reaction in organic chemistry in which an α-diazocarbonyl compound is converted into a ketene by loss of dinitrogen with accompanying 1,2-rearrangement. The Wolff rearrangement yields a ketene as an intermediate product, which can undergo nucleophilic attack with weakly acidic nucleophiles such as water, alcohols, and amines, to generate carboxylic acid derivatives or undergo [2+2] cycloaddition reactions to form four-membered rings. The mechanism of the Wolff rearrangement has been the subject of debate since its first use. No single mechanism sufficiently describes the reaction, and there are often competing concerted and carbene-mediated pathways; for simplicity, only the textbook, concerted mechanism is shown below. The reaction was discovered by Ludwig Wolff in 1902. The Wolff rearrangement has great synthetic utility due to the accessibility of α-diazocarbonyl compounds, variety of reactions from the ketene intermediate, and stereochemical retention of the migrating group. However, the Wolff rearrangement has limitations due to the highly reactive nature of α-diazocarbonyl compounds, which can undergo a variety of competing reactions.
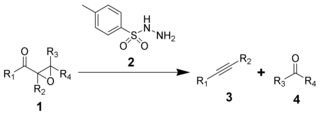
The Eschenmoser fragmentation, first published in 1967, is the chemical reaction of α,β-epoxyketones (1) with aryl sulfonylhydrazines (2) to give alkynes (3) and carbonyl compounds (4). The reaction is named after the Swiss chemist Albert Eschenmoser, who devised it in collaboration with an industrial research group of Günther Ohloff, and applied it to the production of muscone and related macrocyclic musks. The reaction is also sometimes known as the Eschenmoser–Ohloff fragmentation or the Eschenmoser–Tanabe fragmentation as Masato Tanabe independently published an article on the reaction the same year. The general formula of the fragmentation using p-toluenesulfonylhydrazide is:

The Fukuyama indole synthesis is a versatile tin mediated chemical reaction that results in the formation of 2,3-disubstituted indoles. A practical one-pot reaction that can be useful for the creation of disubstituted indoles. Most commonly tributyltin hydride is utilized as the reducing agent, with azobisisobutyronitrile (AIBN) as a radical initiator. Triethylborane can also be used as a radical initiator. The reaction can begin with either an ortho-isocyanostyrene or a 2-alkenylthioanilide derivative, both forming the indole through Radical cyclization via an α-stannoimidoyl radical. The R group can be a range of both basic and acidic sensitive functional groups such as esters, THP ethers, and β-lactams. In addition the reaction is not stereospecific, in that both the cis and trans isoform can be used to obtain the desired product.
Selenoxide elimination is a method for the chemical synthesis of alkenes from selenoxides. It is most commonly used to synthesize α,β-unsaturated carbonyl compounds from the corresponding saturated analogues. It is mechanistically related to the Cope reaction.

Indole is an aromatic, heterocyclic, organic compound with the formula C8H7N. It has a bicyclic structure, consisting of a six-membered benzene ring fused to a five-membered pyrrole ring. Indole is widely distributed in the natural environment and can be produced by a variety of bacteria. As an intercellular signal molecule, indole regulates various aspects of bacterial physiology, including spore formation, plasmid stability, resistance to drugs, biofilm formation, and virulence. The amino acid tryptophan is an indole derivative and the precursor of the neurotransmitter serotonin.
The Fiesselmann thiophene synthesis is a name reaction in organic chemistry that allows for the generation of 3-hydroxy-2-thiophenecarboxylic acid derivatives from α,β-acetylenic esters with thioglycolic acid and its derivatives under the presence of a base. The reaction was developed by Hans Fiesselmann in the 1950s.
