An enamine is a functional group with the formula R2N−C(R′)=CR″2.[1][2] Enamines are reagents used in organic synthesis and are intermediates in some enzyme-catalyzed reactions.[3]
The word "enamine" is derived from the affix en-, used as the suffix of alkene, and the root amine. This can be compared with enol, which is a functional group containing both alkene (en-) and alcohol (-ol). Enamines are nitrogen analogs of enols.[4]
Enamines are both good nucleophiles and good bases. Their behavior as carbon-based nucleophiles is explained with reference to the following resonance structures.
Resonance structures for an enamine
Formation
Condensation to give an enamine.
Enamines can be easily produced from commercially available starting reagents. Commonly enamines are produced by condensation of secondary amines with ketones and aldehydes..[3][6] The condensing ketone and aldehyde must contain an α-hydrogen. The associated equations for enamine formation follow:
In some cases, acid-catalysts are employed. Acid catalysis is not always required, if the pKaH of the reacting amine is sufficiently high (for example, pyrrolidine, which has a pKaH of 11.26). If the pKaH of the reacting amine is low, however, then acid catalysis is required through both the addition and the dehydration steps.[7] Common dehydrating agents include MgSO4 and Na2SO4.[8]
Methyl ketone self-condensation is a side-reaction which can be avoided through the addition of TiCl4[9] into the reaction mixture (to act as a water scavenger).[8][10]
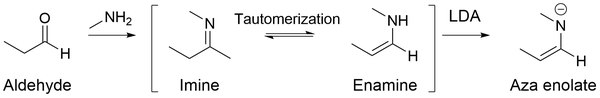
Primary amines are usually not used for enamine synthesis.[11] Instead, such reactions give imines:
Selected bond distances (picometers) in an enamine. Atoms in red are nearly coplanar.
As shown by X-ray crystallography, the C3NC2 portion of enamines is close to planar. This arrangement reflects the sp2hybridization of the C=CN core.
E vs Z geometry affects the reactivity of enamines.[8]
Reactions
Enamines are nucleophiles. Ketone enamines are more nucleophilic than their aldehyde counterparts.[13]
Compared to their enolate counterparts, their alkylations often proceed with fewer side reactions. Cyclic ketone enamines follow a reactivity trend where the five membered ring is the most reactive due to its maximally planar conformation at the nitrogen, following the trend 5>8>6>7 (the seven membered ring being the least reactive). This trend has been attributed to the amount of p-character on the nitrogen lone pair orbital - the higher p character corresponding to a greater nucleophilicity because the p-orbital would allow for donation into the alkene π- orbital. Analogously, if the N lone pair participates in stereoelectronic interactions on the amine moiety, the lone pair will pop out of the plane (will pyramidalize) and compromise donation into the adjacent π C-C bond.[14]
Alkylation and acylation
Alkylation is the predominant reaction sought with enamines. When treated with alkyl halides enamines give the alkylated iminium salts, which then can be hydrolyzes to regenerate a ketone (a starting material in enamine synthesis):
R2N−CH=CHR' + R"X → [R2N+=CH−CHR'R"]X− (alkylation of enamine)
[R2N+=CH−CHR'R"]+X− + H2O → R2NH + R'R"CHCHO (hydrolysis of the resulting iminium salt, giving a 2-alkylated aldehyde)
Owing to the pioneering work by Gilbert Stork, this reaction is sometimes referred to as the Stork enamine alkylation. Analogously, this reaction can be used as an effective means of acylation. A variety of alkylating and acylating agents including benzylic, allylic halides can be used in this reaction.[15]
Similar to their alkylation, enamines can be acylated. Hydrolysis of this acylated imine forms a 1,3-dicarbonyl.[16][11]
R2N−CH=CHR' + R"COCl → [R2N+=CH−CHR'C(O)R"]Cl− (acylation of enamine)
[R2N+=CH−CHR'C(O)R"]+Cl + H2O → R2NH + O=C(H)CH(R')CR"=O (hydrolysis of the resulting acyl iminium salt, giving a C-acylated aldehyde)
Halogenation
Chlorination of enamines followed by hydrolysis gives α-halo ketones and aldehydes:
R2NCH=CHR' + Cl2 → [R2N+=CH−CHR'CCl]Cl− (chlorination of enamine)
[R2N+=CH−CHR'Cl]Cl− + H2O → R2NH + R'CH(Cl)CHO (hydrolysis of chloroiminium, giving a chloroaldehyde)
In addition to chlorination, bromination and even iodination have been demonstrated.[17]
Oxidative coupling
Enamines can be efficiently cross-coupled with enol silanes through treatment with ceric ammonium nitrate.[18] Oxidative dimerization of aldehydes in the presence of amines proceeds through the formation of an enamine followed by a final pyrrole formation.[19] This method for symmetric pyrrole synthesis was developed in 2010 by the Jia group, as a valuable new pathway for the synthesis of pyrrole-containing natural products.[20]
Annulation
Enamines chemistry has been implemented for the purposes of producing a one-pot enantioselective version of the Robinson annulation. The Robinson annulation, published by Robert Robinson in 1935, is a base-catalyzed reaction that combines a ketone and a methyl vinyl ketone (commonly abbreviated to MVK) to form a cyclohexenone fused ring system. This reaction may be catalyzed by proline to proceed through chiral enamine intermediates which allow for good stereoselectivity.[21] This is important, in particular in the field of natural product synthesis, for example, for the synthesis of the Wieland-Miescher ketone – a vital building block for more complex biologically active molecules.[22][23]
Metalloenamines
Lithiated enamines (also known as aza enolates, imine anions, enamides, or metallated Schiff bases) are nitrogen analogues to enolates,[24] formed when imines get treated with strong bases such as LiNR2:
That reaction is one of the key steps in the synthesis of the Oulema melanopus' male aggression pheromone:[30]
Most prominently, these reactions have allowed for asymmetric alkylations of ketones through transformation to chiral intermediate metalloenamines.[31]
Nature processes (makes and degrades) sugars using enzymes called aldolases. These enzymes act by reversible formation of enamines.[32][33]
Further reading
Early literature of historic interest:
the term "enamine" is coined: Wittig, Georg; Blumenthal, Hermann (1927). "Über die Einwirkung von Ammoniak und Ammoniak-Derivaten auf o -Acetylaceto-phenole". Berichte der Deutschen Chemischen Gesellschaft (A and B Series). 60 (5): 1085–1094. doi:10.1002/cber.19270600515.
↑ Capon, Brian; Wu, Zhen Ping (April 1990). "Comparison of the tautomerization and hydrolysis of some secondary and tertiary enamines". The Journal of Organic Chemistry. 55 (8): 2317–2324. doi:10.1021/jo00295a017.
↑ White, William Andrew; Weingarten, Harold (January 1967). "A versatile new enamine synthesis". The Journal of Organic Chemistry. 32 (1): 213–214. doi:10.1021/jo01277a052.
↑ Li, Q; Fan, A; Lu, Z; Cui, Y; Lin, W; Jia, Y (2010). "One-pot AgOAc-mediated synthesis of polysubstituted pyrroles from primary amines and aldehydes: application to the total synthesis of purpurone". Organic Letters. 12 (18): 4066–4069. doi:10.1021/ol101644g. PMID20734981.
↑ Carey, Francis A. (2007). Advanced organic chemistry. Part B, Reactions and synthesis (5thed.). New York, NY: Springer. pp.46–47. ISBN978-0-387-68350-8.
↑ Clayden, Jonathan (2012). Organic chemistry (2nded.). Oxford: Oxford University Press. pp.465, 593–594. ISBN978-0-19-927029-3.
↑ Hudrlik, Paul F.; Wan, Chung-Nan (October 1975). "Reactions of oxetane with imine salts derived from cyclohexanone". The Journal of Organic Chemistry. 40 (20): 2963–2965. doi:10.1021/jo00908a027.
↑ Chevalley, Alice; Férézou, Jean-Pierre (2012). "One-pot formation of aza-enolates from secondary amines and condensation to esters and alkyl bromides". Tetrahedron. 68 (29): 5882–5889. doi:10.1016/j.tet.2012.04.105.
↑ Meyers, A. I.; Williams, Donald R. (August 1978). "Asymmetric alkylation of acyclic ketones via chiral metallo enamines. Effect of kinetic vs. thermodynamic metalations". The Journal of Organic Chemistry. 43 (16): 3245–3247. doi:10.1021/jo00410a034.
↑ Notz, Wolfgang; Tanaka, Fujie; Barbas, Carlos F. (2004). "Enamine-Based Organocatalysis with Proline and Diamines: The Development of Direct Catalytic Asymmetric Aldol, Mannich, Michael, and Diels−Alder Reactions". Accounts of Chemical Research. 37 (8): 580–591. doi:10.1021/ar0300468. PMID15311957.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.