Keto–enol tautomerism refers to a chemical equilibrium between a "keto" form (a carbonyl, named for the common ketone case) and an enol. The tautomeric interconversion involves hydrogen atom movement and the reorganisation of bonding electrons.[1]
Many kinds of enols are known, but very few are stable compounds.[2] However, deprotonation of organic carbonyls gives enolate anions, which are important in organic reaction strategies as a strong nucleophile.
The process does not occur intramolecularly, but requires participation of solvent or other mediators.[citation needed]
Strictly speaking, the conversion is a keto-enol tautomerism only in the case of ketones (neither R nor R′ hydrogen). But this name is often more generally applied to all such tautomerizations.
The keto-enol equilibrium involves movement of a double bond. If the α position of an enol is substituted (i.e., not a methyl ketone), then it is prochiral, forming a new stereocenter when in keto form. Conversely, enolization racemizes that stereocenter.[citation needed]
Usually the tautomerization equilibrium constant is so small that the enol is undetectable spectroscopically. In the equilibrium between vinyl alcohol and acetaldehyde, K=[enol]/[keto]≈5.8×10−7.[3]
The terminus of the double bond in enols is nucleophilic, a property enhanced in the case of enolate anions.[4][5] However, enolates protonate reversibly at the oxygen much faster than equilibrate to the ketone/aldehyde/etc.[6] As many organic syntheses involve the controlled formation and reaction of enolates, enols appear transiently in great quantities during quenching.[4][5]
Stable enols
Diaryl-substitution stabilizes some enols.
Enols can be stabilized through vinylogy. Thus, very stable enols are phenols.[8]
In compounds with two (or more) carbonyls, the enol form is also stabilized through intramolecular hydrogen bonding[9] and becomes dominant. The behavior of 2,4-pentanedione illustrates this effect:[10]
Phenols represent a kind of enol. For some phenols and related compounds, the keto tautomer plays an important role. Many of the reactions of resorcinol and phloroglucinol involve the keto tautomers, for example. Naphthalene-1,4-diol exists in observable equilibrium with the diketone tetrahydronaphthalene-1,4-dione.[11]
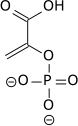
The high phosphate-transfer potential of phosphoenolpyruvate results from the fact that the phosphorylated compound is "trapped" in the less thermodynamically favorable enol form, whereas after dephosphorylation it can assume the keto form.[citation needed]
Enediols are alkenes with a hydroxyl group on each carbon of the C=C double bond. Normally such compounds are disfavored components in equilibria with acyloins. One special case is catechol, where the C=C subunit is part of an aromatic ring. In some other cases however, enediols are stabilized by flanking carbonyl groups. These stabilized enediols are called reductones. Such species are important in glycochemistry, e.g., the Lobry de Bruyn–Van Ekenstein transformation.[13]
Conversion of ascorbic acid (vitamin C) to an enolate. Enediol at left, enolate at right, showing movement of electron pairs resulting in deprotonation of the stable parent enediol. A distinct, more complex chemical system, exhibiting the characteristic of vinylogy.
12Guthrie, J. Peter; Povar, Igor (2013). "Equilibrium constants for enolization in solution by computation alone". Journal of Physical Organic Chemistry. 26 (12): 1077–1083. doi:10.1002/poc.3168 See column "pKExpt E" in Table1; values there are negative decimal logarithms of values here.
↑Manbeck, Kimberly A.; Boaz, Nicholas C.; Bair, Nathaniel C.; Sanders, Allix M. S.; Marsh, Anderson L. (2011). "Substituent Effects on Keto–Enol Equilibria Using NMR Spectroscopy". J. Chem. Educ.88 (10): 1444–1445. Bibcode:2011JChEd..88.1444M. doi:10.1021/ed1010932.
↑Kündig, E. Peter; Enríquez García, Alvaro; Lomberget, Thierry; Bernardinelli, Gérald (2006). "Rediscovery, Isolation, and Asymmetric Reduction of 1,2,3,4-Tetrahydronaphthalene-1,4-dione and Studies of Its [Cr(CO)3] Complex". Angewandte Chemie International Edition. 45 (1): 98–101. doi:10.1002/anie.200502588. PMID16304647.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.