
In organic chemistry, enolates are the organic anions derived from the deprotonation of carbonyl (RR'C=O) compounds. Rarely isolated, they are widely used as reagents in the synthesis of organic compounds. [1] [2] [3] [4]
In organic chemistry, enolates are the organic anions derived from the deprotonation of carbonyl (RR'C=O) compounds. Rarely isolated, they are widely used as reagents in the synthesis of organic compounds. [1] [2] [3] [4]

Enolate anions are electronically related to allyl anions. The anionic charge is delocalized over the oxygen and the two carbon sites. Thus they have the character of both an alkoxide and a carbanion. [5]

Although enolate salts are often drawn as simple ion pairs, in fact they adopt complicated structures often featuring aggregates. [7]
Carbonyl compounds with an α hydrogen atom deprotonate to give enolates: [8] [9]

Base mediates the process, but Lewis acidity plays a key role in stabilizing the product. Often the (weak) Lewis acid is simply the alkali counterion to an Arrhenius base (1.); unsurprisingly, reactivity with such salts varies from lithium to cesium. Alternatively, enolates can be generated from a molecular Lewis acid and a weak Brønsted base (a frustrated Lewis pair; 2.):

Most substrates have multiple α hydrogen atoms, and in principle could give multiple enolate isomers. For example, with methylcyclohexanone:

However, reaction conditions can control both the resulting enolate's regio- [11] and stereochemistry. [12] This provides one of the best understood synthetic strategies to introduce chemical complexity in total syntheses.[ citation needed ]
Alternatively, an enone can serve as a protecting group, masking a specific enol. [13] Reaction with a hydride or dissolving-metal reduction then forms an enol, as in this total synthesis of progesterone: [14]

The kinetic-versus-thermodynamic distinction is key to regiocontrol during deprotonation. Substitution improves alkene thermodynamics through additional hyperconjugation, but hinders initial proton loss. In the methylcyclohexanone example above, the trisubstituted enolate deprotonates more quickly: it is the kinetic enolate. The tetrasubstituted enolate is more stable, and dominant in thermodynamic equilibrium.
Base strength determines the regioisomeric ratio. With strong bases and weak Lewis acids, deprotonation is quantitative and irreversible, trapping the kinetic enolate. Typically kinetic enolates are generated using lithium diisopropylamide (LDA), often in slight excess and at low temperature. [15] Weaker alkoxide bases and stronger Lewis acids instead reversibly deprotonate the substrate, affording thermodynamically-favored enolates.
Most enolization conditions give Z enolates from ketones and E enolates from esters, but HMPA is known to reverse the stereoselectivity of deprotonation.

Likewise different Lewis acids give different enolate geometries: [12]

The Ireland model attempts to rationalize stereoselection [16] [17] [18] [19] with a six-membered, cyclic, [20] monomeric transition state proposal. For deprotonation to occur, an α C-H σ bond must overlap the π* orbital of the carbonyl:
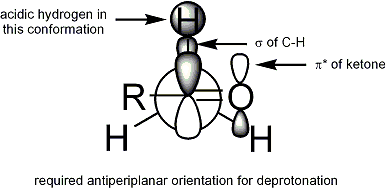
In the Ireland model, the larger substituent on the electrophile (for the ester above, methyl) adopts an equatorial disposition in the transition state, leading to a preference for E enolates.

The Ireland model fails often. It is not known when, if ever, the intermediates are monomeric and cyclic like the model proposes.
Powerful nucleophiles, enolates react with a variety of electrophiles at oxygen and carbon. Controlling which atom enolates react at has drawn much attention. Reaction at carbon is thermodynamically favored. Kinetically, the negative charge in enolates is concentrated on the oxygen. However, the oxygen center is also highly solvated, which can lead to alkylation at carbon. [21]
Reaction at oxygen traps the enolate as a (silyl) [22] enol ether [23] or ester. [24] Such species can be carried through other transformations relatively inertly, but then are released in the presence of Lewis acids (the Mukaiyama aldol reaction):

Other important electrophiles are aldehydes/ketones (the aldol reaction) and Michael acceptors. [25]
