Lithium diisopropylamide (commonly abbreviated LDA) is a chemical compound with the molecular formulaLiN(CH(CH3)2)2. It is used as a strong base and has been widely utilized due to its good solubility in non-polar organic solvents and non-nucleophilic nature. It is a colorless solid, but is usually generated and observed only in solution. It was first prepared by Hamell and Levine in 1950 along with several other hindered lithium diorganylamides to effect the deprotonation of esters at the α position without attack of the carbonyl group.[2]
When dissociated, the diisopropylamide anion can become protonated to form diisopropylamine. Diisopropylamine has a pKa value of 36. Therefore, its conjugate base is suitable for the deprotonation of compounds with greater acidity, importantly, such weakly acidic compounds (carbon acids) of the type HC(Z)R2, where Z = C(O)R', C(O)OR' or CN. Conventional protic functional groups such as alcohols and carboxylic acids are readily deprotonated.
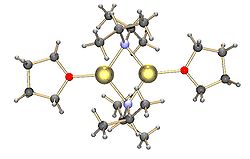
Like most organolithium reagents, LDA is not a salt, but is highly polar. It forms aggregates in solution, with the extent of aggregation depending on the nature of the solvent. In THF its structure is primarily that of a solvated dimer.[4][5] In nonpolar solvents such as toluene, it forms a temperature-dependent oligomer equilibrium. At room temperature trimers and tetramers are the most likely structures. With decreasing temperature the aggregation extends to pentameric and higher oligomeric structures.[6]
Solid LDA is pyrophoric,[7] but its solutions are generally not. As such it is commercially available as a solution in polar aprotic solvents such as THF and ether; however, for small scale use (less than 50mmol), it is common and more cost effective to prepare LDA in situ.
Deprotonation using LDA.
Kinetic vs thermodynamic bases
The deprotonation of carbon acids can proceed with either kinetic or thermodynamic reaction control. Kinetic controlled deprotonation requires a base that is sterically hindered and strong enough to remove the proton irreversibly. For example, in the case of phenylacetone, deprotonation can produce two different enolates. LDA has been shown to deprotonate the methyl group, which is the kinetic course of the deprotonation. To ensure the production of the kinetic product, a slight excess (1.1 equiv) of lithium diisopropylamide is used, and the ketone is added to the base at –78°C. Because the ketone is quickly and quantitatively converted to the enolate and base is present in excess at all times, the ketone is unable to act as a proton shuttle to catalyze the gradual formation of the thermodynamic product. A weaker base such as an alkoxide, which reversibly deprotonates the substrate, affords the more thermodynamically stable benzylic enolate. An alternative to the weaker base is to use a strong base which is present at a lower concentration than the ketone. For instance, with a slurry of sodium hydride in THF or dimethylformamide (DMF), the base only reacts at the solution–solid interface. A ketone molecule might be deprotonated at the kinetic site. This enolate may then encounter other ketones and the thermodynamic enolate will form through the exchange of protons, even in an aprotic solvent which does not contain hydronium ions.
LDA can, however, act as a nucleophile under certain conditions.
↑Hamell, Matthew; Levine, Robert (1950). "Condensations Effected by the Alkali Amides. IV. The Reactions of Esters with Lithium Amide and Certain Substituted Lithium Amides1". The Journal of Organic Chemistry. 15: 162–168. doi:10.1021/jo01147a026.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.