Justus von Liebig's synthesis of oxamide from dicyan and water represents the first organocatalytic reaction, with acetaldehyde further identified as the first discovered pure "organocatalyst", which act similarly to the then-named "ferments", now known as enzymes.
In organic chemistry, organocatalysis is a form of catalysis in which the rate of a chemical reaction is increased by an organic catalyst. This "organocatalyst" consists of carbon, hydrogen, sulfur and other nonmetal elements found in organic compounds.[3][4][5][6][7][8] Because of their similarity in composition and description, they are often mistaken as a misnomer for enzymes due to their comparable effects on reaction rates and forms of catalysis involved.
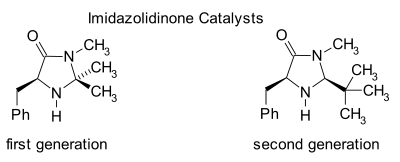
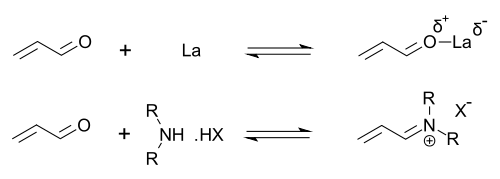
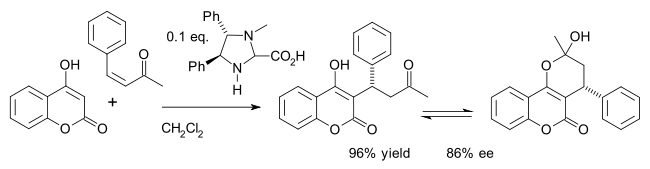
Organocatalysts which display secondary amine functionality can be described as performing either enamine catalysis (by forming catalytic quantities of an active enamine nucleophile) or iminium catalysis (by forming catalytic quantities of an activated iminium electrophile). This mechanism is typical for covalent organocatalysis. Covalent binding of substrate normally requires high catalyst loading (for proline-catalysis typically 20–30mol%). Noncovalent interactions such as hydrogen-bonding facilitates low catalyst loadings (down to 0.001mol%).
Organocatalysis offers several advantages. There is no need for metal-based catalysis thus making a contribution to green chemistry. In this context, simple organic acids have been used as catalyst for the modification of cellulose in water on multi-ton scale.[9] When the organocatalyst is chiral an avenue is opened to asymmetric catalysis; for example, the use of proline in aldol reactions is an example of chirality and green chemistry.[10] Organic chemists David MacMillan and Benjamin List were both awarded the 2021 Nobel Prize in chemistry for their work on asymmetric organocatalysis.[11]
Introduction
Regular achiral organocatalysts are based on nitrogen such as piperidine used in the Knoevenagel condensation.[12]DMAP used in esterifications[13] and DABCO used in the Baylis-Hillman reaction.[14]Thiazolium salts are employed in the Stetter reaction.[15] These catalysts and reactions have a long history but current interest in organocatalysis is focused on asymmetric catalysis with chiral catalysts, called asymmetric organocatalysis or enantioselective organocatalysis. A pioneering reaction developed in the 1970s is called the Hajos–Parrish–Eder–Sauer–Wiechert reaction. Between 1968 and 1997, there were only a few reports of the use of small organic molecules as catalysts for asymmetric reactions (the Hajos–Parrish reaction probably being the most famous), but these chemical studies were viewed more as unique chemical reactions than as integral parts of a larger, interconnected field.[16]
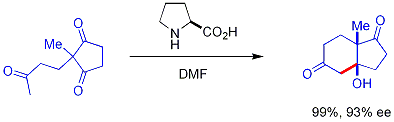
In this reaction, naturally occurring chiral proline is the chiral catalyst in an aldol reaction. The starting material is an achiral triketone and it requires just 3% of proline to obtain the reaction product, a ketol in 93% enantiomeric excess. This is the first example of an amino acid-catalyzed asymmetric aldol reaction.[17][18]
The asymmetric synthesis of the Wieland-Miescher ketone (1985) is also based on proline and another early application was one of the transformations in the total synthesis of Erythromycin by Robert B. Woodward (1981).[19] A mini-review digest article focuses on selected recent examples of total synthesis of natural and pharmaceutical products using organocatalytic reactions.[20]
Many chiral organocatalysts are an adaptation of chiral ligands (which together with a metal center also catalyze asymmetric reactions) and both concepts overlap to some degree.
A breakthrough in the field of organocatalysis came in 1997 when Yian Shi reported the first general, highly enantioselective organocatalytic reaction with the catalytic asymmetric epoxidation of trans- and trisubstituted olefins with chiral dioxiranes.[21] Since that time, several different types of reactions have been developed.
Organocatalyst classes
Organocatalysts for asymmetric synthesis can be grouped in several classes:
A large group of organocatalysts incorporate the urea or the thiourea moiety. These catalytically effective (thio)urea derivatives termed (thio)urea organocatalysts provide explicit double hydrogen-bonding interactions to coordinate and activate H-bond accepting substrates.[29]
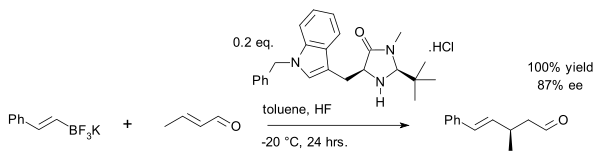
Their current uses are restricted to asymmetric multicomponent reactions, including those involving Michael addition, asymmetric multicomponent reactions for the synthesis of spirocycles, asymmetric multicomponent reactions involving acyl Strecker reactions, asymmetric Petasis reactions, asymmetric Biginelli reactions, asymmetric Mannich reactions, asymmetric aza-Henry reactions, and asymmetric reductive coupling reactions.[30]
↑ W. Langenbeck (1929). "Über organische Katalysatoren. III. Die Bildung von Oxamid aus Dicyan bei Gegenwart von Aldehyden". Liebigs Ann. 469: 16–25. doi:10.1002/jlac.19294690103.
↑ Peter I. Dalko; Lionel Moisan (2004). "In the Golden Age of Organocatalysis". Angew. Chem. Int. Ed. 43 (39): 5138–5175. doi:10.1002/anie.200400650. PMID15455437.
↑ Matthew J. Gaunt; Carin C.C. Johansson; Andy McNally; Ngoc T. Vo (2007). "Enantioselective organocatalysis". Drug Discovery Today. 12 (1/2): 8–27. doi:10.1016/j.drudis.2006.11.004. PMID17198969.
↑ Dieter Enders; Christoph Grondal; Matthias R. M. Hüttl (2007). "Asymmetric Organocatalytic Domino Reactions". Angew. Chem. Int. Ed. 46 (10): 1570–1581. doi:10.1002/anie.200603129. PMID17225236.
↑ International Patent WO 2006068611 A1 20060629 " Direct Homogeneous and Heterogeneous Organic Acid and Amino Acid-Catalyzed Modification of Amines and Alcohols" Inventors: Armando Córdova, Stockholm, Sweden; Jonas Hafrén, Stockholm, Sweden.
↑ Example 4 in U.S. Patent 3,975,440 August 17, 1976, Filed Dec. 9, 1970 Zoltan G. Hajos and David R. Parrish.
↑ List, B. (2010). "Emil Knoevenagel and the Roots of Aminocatalysis". Angewandte Chemie International Edition in English. 49 (10): 1730–1734. doi:10.1002/anie.200906900. PMID20175175.
↑ Neises, Bernhard; Steglich, Wolfgang (July 1978). "Simple Method for the Esterification of Carboxylic Acids". Angewandte Chemie International Edition in English. 17 (7): 522–524. doi:10.1002/anie.197805221.
↑ Basavaiah, Deevi; Rao, Anumolu Jaganmohan; Satyanarayana, Tummanapalli (March 2003). "Recent Advances in the Baylis−Hillman Reaction and Applications". Chemical Reviews. 103 (3): 811–892. doi:10.1021/cr010043d. PMID12630854.
↑ Z. G. Hajos, D. R. Parrish, German Patent DE 2102623 1971
↑ Zoltan G. Hajos; David R. Parrish (1974). "Asymmetric synthesis of bicyclic intermediates of natural product chemistry". J. Org. Chem. 39 (12): 1615–1621. doi:10.1021/jo00925a003.
↑ R. B. Woodward; E. Logusch; K. P. Nambiar; K. Sakan; D. E. Ward; B. W. Au-Yeung; P. Balaram; L. J. Browne; etal. (1981). "Asymmetric total synthesis of erythromcin. 1. Synthesis of an erythronolide A secoacid derivative via asymmetric induction". J. Am. Chem. Soc. 103 (11): 3210–3213. Bibcode:1981JAChS.103.3210W. doi:10.1021/ja00401a049.
↑ Bertelsen, Søren (2009). "Organocatalysis—after the gold rush". Chemical Society Reviews. 38 (8): 2178–89. doi:10.1039/b903816g. PMID19623342.
↑ Gaunt, M. J.; Johansson, C. C. C.; McNally, A.; Vo, N. T. (2007). "Enantioselective organocatalysis". Drug Discovery Today. 12 (1–2): 8–27. doi:10.1016/j.drudis.2006.11.004. PMID17198969.
↑ Kucherenko, A. S.; Siyutkin, D. E.; Maltsev, O. V.; Kochetkov, S. V.; Zlotin, S. G. (2013). "Asymmetric organocatalysis: From proline to highly efficient immobilized organocatalysts". Russian Chemical Bulletin. 61 (7): 1313. doi:10.1007/s11172-012-0177-4. S2CID93168492.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.