The aldol reaction is a reaction in organic chemistry that combines two carbonyl compounds to form a new β-hydroxy carbonyl compound. Its simplest form might involve the nucleophilic addition of an enolized ketone to another:

An enamine is an unsaturated compound derived by the condensation of an aldehyde or ketone with a secondary amine. Enamines are versatile intermediates.

In organometallic chemistry, organolithium reagents are chemical compounds that contain carbon–lithium (C–Li) bonds. These reagents are important in organic synthesis, and are frequently used to transfer the organic group or the lithium atom to the substrates in synthetic steps, through nucleophilic addition or simple deprotonation. Organolithium reagents are used in industry as an initiator for anionic polymerization, which leads to the production of various elastomers. They have also been applied in asymmetric synthesis in the pharmaceutical industry. Due to the large difference in electronegativity between the carbon atom and the lithium atom, the C−Li bond is highly ionic. Owing to the polar nature of the C−Li bond, organolithium reagents are good nucleophiles and strong bases. For laboratory organic synthesis, many organolithium reagents are commercially available in solution form. These reagents are highly reactive, and are sometimes pyrophoric.

In organic chemistry, the ene reaction is a chemical reaction between an alkene with an allylic hydrogen and a compound containing a multiple bond, in order to form a new σ-bond with migration of the ene double bond and 1,5 hydrogen shift. The product is a substituted alkene with the double bond shifted to the allylic position.

In organic chemistry, enolates are organic anions derived from the deprotonation of carbonyl compounds. Rarely isolated, they are widely used as reagents in the synthesis of organic compounds.

Organoboron chemistry or organoborane chemistry studies organoboron compounds, also called organoboranes. These chemical compounds combine boron and carbon; typically, they are organic derivatives of borane (BH3), as in the trialkyl boranes.
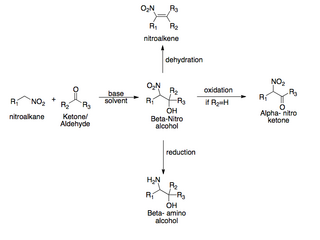
The Henry reaction is a classic carbon–carbon bond formation reaction in organic chemistry. Discovered in 1895 by the Belgian chemist Louis Henry (1834–1913), it is the combination of a nitroalkane and an aldehyde or ketone in the presence of a base to form β-nitro alcohols. This type of reaction is also referred to as a nitroaldol reaction. It is nearly analogous to the aldol reaction that had been discovered 23 years prior that couples two carbonyl compounds to form β-hydroxy carbonyl compounds known as "aldols". The Henry reaction is a useful technique in the area of organic chemistry due to the synthetic utility of its corresponding products, as they can be easily converted to other useful synthetic intermediates. These conversions include subsequent dehydration to yield nitroalkenes, oxidation of the secondary alcohol to yield α-nitro ketones, or reduction of the nitro group to yield β-amino alcohols.

In stereochemistry, a chiral auxiliary is a stereogenic group or unit that is temporarily incorporated into an organic compound in order to control the stereochemical outcome of the synthesis. The chirality present in the auxiliary can bias the stereoselectivity of one or more subsequent reactions. The auxiliary can then be typically recovered for future use.

Nucleophilic conjugate addition is a type of organic reaction. Ordinary nucleophilic additions or 1,2-nucleophilic additions deal mostly with additions to carbonyl compounds. Simple alkene compounds do not show 1,2 reactivity due to lack of polarity, unless the alkene is activated with special substituents. With α,β-unsaturated carbonyl compounds such as cyclohexenone it can be deduced from resonance structures that the β position is an electrophilic site which can react with a nucleophile. The negative charge in these structures is stored as an alkoxide anion. Such a nucleophilic addition is called a nucleophilic conjugate addition or 1,4-nucleophilic addition. The most important active alkenes are the aforementioned conjugated carbonyls and acrylonitriles.

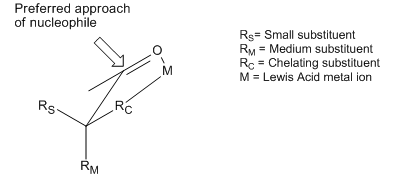
The Bürgi–Dunitz angle is one of two angles that fully define the geometry of "attack" of a nucleophile on a trigonal unsaturated center in a molecule, originally the carbonyl center in an organic ketone, but now extending to aldehyde, ester, and amide carbonyls, and to alkenes (olefins) as well. The angle was named after crystallographers Hans-Beat Bürgi and Jack D. Dunitz, its first senior investigators.
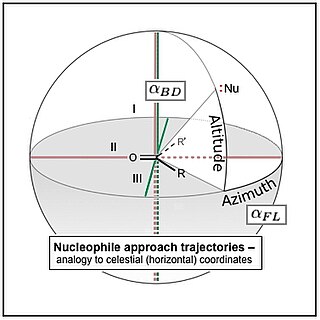
The Flippin–Lodge angle is one of two angles used by organic and biological chemists studying the relationship between a molecule's chemical structure and ways that it reacts, for reactions involving "attack" of an electron-rich reacting species, the nucleophile, on an electron-poor reacting species, the electrophile. Specifically, the angles—the Bürgi–Dunitz, , and the Flippin–Lodge, —describe the "trajectory" or "angle of attack" of the nucleophile as it approaches the electrophile, in particular when the latter is planar in shape. This is called a nucleophilic addition reaction and it plays a central role in the biological chemistry taking place in many biosyntheses in nature, and is a central "tool" in the reaction toolkit of modern organic chemistry, e.g., to construct new molecules such as pharmaceuticals. Theory and use of these angles falls into the areas of synthetic and physical organic chemistry, which deals with chemical structure and reaction mechanism, and within a sub-specialty called structure correlation.

Organoindium chemistry is the chemistry of compounds containing In-C bonds. The main application of organoindium chemistry is in the preparation of semiconducting components for microelectronic applications. The area is also of some interest in organic synthesis. Most organoindium compounds feature the In(III) oxidation state, akin to its lighter congeners Ga(III) and B(III).
The α-ketol rearrangement is the acid-, base-, or heat-induced 1,2-migration of an alkyl or aryl group in an α-hydroxy ketone or aldehyde to give an isomeric product.
Organostannane addition is reaction involving the nucleophilic addition of an allyl-, allenyl-, or propargyl-stannane to an aldehyde, imine, or a ketone. This reaction is widely used for carbonyl allylation.
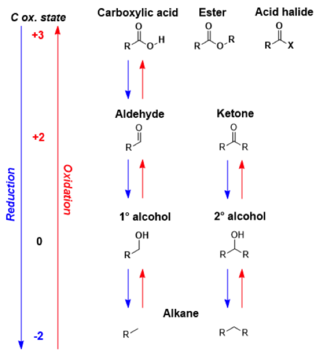
In organic chemistry, carbonyl reduction is the conversion of any carbonyl group, usually to an alcohol. It is a common transformation that is practiced in many ways. Ketones, aldehydes, carboxylic acids, esters, amides, and acid halides - some of the most pervasive functional groups, -comprise carbonyl compounds. Carboxylic acids, esters, and acid halides can be reduced to either aldehydes or a step further to primary alcohols, depending on the strength of the reducing agent. Aldehydes and ketones can be reduced respectively to primary and secondary alcohols. In deoxygenation, the alcohol group can be further reduced and removed altogether by replacement with H.
Reductions with metal alkoxyaluminium hydrides are chemical reactions that involve either the net hydrogenation of an unsaturated compound or the replacement of a reducible functional group with hydrogen by metal alkoxyaluminium hydride reagents.
In organic chemistry, the Baylis–Hillman, Morita–Baylis–Hillman, or MBH reaction is a carbon-carbon bond-forming reaction between an activated alkene and a carbon electrophile in the presence of a nucleophilic catalyst, such as a tertiary amine or phosphine. The product is densely functionalized, joining the alkene at the α-position to a reduced form of the electrophile.
In organic chemistry, Lewis acid catalysis is the use of metal-based Lewis acids as catalysts for organic reactions. The acids act as an electron pair acceptor to increase the reactivity of a substrate. Common Lewis acid catalysts are based on main group metals such as aluminum, boron, silicon, and tin, as well as many early and late d-block metals. The metal atom forms an adduct with a lone-pair bearing electronegative atom in the substrate, such as oxygen, nitrogen, sulfur, and halogens. The complexation has partial charge-transfer character and makes the lone-pair donor effectively more electronegative, activating the substrate toward nucleophilic attack, heterolytic bond cleavage, or cycloaddition with 1,3-dienes and 1,3-dipoles.

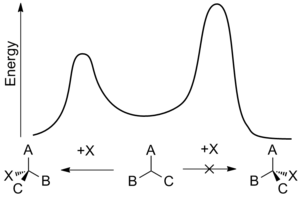
Dynamic kinetic resolution in chemistry is a type of kinetic resolution where 100% of a racemic compound can be converted into an enantiopure compound. It is applied in asymmetric synthesis. Asymmetric synthesis has become a much explored field due to the challenge of creating a compound with a single 3D structure. Even more challenging is the ability to take a racemic mixture and have only one chiral product left after a reaction. One method that has become an exceedingly useful tool is dynamic kinetic resolution (DKR). DKR utilizes a center of a particular molecule that can be easily epimerized so that the (R) and (S) enantiomers can interconvert throughout the reaction process. At this point the catalyst can selectively lower the transition state energy of a single enantiomer, leading to almost 100% yield of one reaction pathway over the other. The figure below is an example of an energy diagram for a compound with an (R) and (S) isomer.
In organic chemistry, the Cieplak effect is a predictive model to rationalize why nucleophiles preferentially add to one face of a carbonyl over another. Proposed by Andrzej Stanislaw Cieplak in 1980, it correctly predicts results that could not be justified by the other standard models at the time, such as the Cram and Felkin–Anh models. In the Cieplak model, electrons from a neighboring bond delocalize into the forming carbon–nucleophile (C–Nuc) bond, lowering the energy of the transition state and accelerating the rate of reaction. Whichever bond can best donate its electrons into the C–Nuc bond determines which face of the carbonyl the nucleophile will add to. The nucleophile may be any of a number of reagents, most commonly organometallic or reducing agents. The Cieplak effect is subtle, and often competes with sterics, solvent effects, counterion complexation of the carbonyl oxygen, and other effects to determine product distribution. Subsequent work has questioned its legitimacy.