Stereogenic group placed on a molecule to encourage stereoselectivity in reactions
In stereochemistry, a chiral auxiliary is a stereogenic group or unit that is temporarily incorporated into an organic compound in order to control the stereochemical outcome of the synthesis.[1][2] The chirality present in the auxiliary can bias the stereoselectivity of one or more subsequent reactions. The auxiliary can then be typically recovered for future use.
General scheme for employing a chiral auxiliary in asymmetric synthesis
Most biological molecules and pharmaceutical targets exist as one of two possible enantiomers; consequently, chemical syntheses of natural products and pharmaceutical agents are frequently designed to obtain the target in enantiomerically pure form.[3] Chiral auxiliaries are one of many strategies available to synthetic chemists to selectively produce the desired stereoisomer of a given compound.[4]
Chiral auxiliaries were introduced by Elias James Corey in 1975[5] with chiral 8-phenylmenthol and by Barry Trost in 1980 with chiral mandelic acid. The menthol compound is difficult to prepare and as an alternative trans-2-phenyl-1-cyclohexanol was introduced by J. K. Whitesell in 1985.
Asymmetric synthesis
Chiral auxiliaries are incorporated into synthetic routes to control the absolute configuration of stereogenic centers. David A. Evans' synthesis of the macrolide cytovaricin, considered a classic, utilizes oxazolidinone chiral auxiliaries for one asymmetric alkylation reaction and four asymmetric aldol reactions, setting the absolute stereochemistry of nine stereocenters.[6]
Cytovaricin, synthesized by D. A. Evans in 1990. Blue and red bonds indicate stereocenters set with the use of chiral auxiliaries.
A typical auxiliary-guided stereoselective transformation involves three steps: first, the auxiliary is covalently coupled to the substrate; second, the resulting compound undergoes one or more diastereoselective transformations; and finally, the auxiliary is removed under conditions that do not cause racemization of the desired products.[4] The cost of employing stoichiometric auxiliary and the need to spend synthetic steps appending and removing the auxiliary make this approach appear inefficient. However, for many transformations, the only available stereoselective methodology relies on chiral auxiliaries. In addition, transformations with chiral auxiliaries tend to be versatile and very well-studied, allowing the most time-efficient access to enantiomerically pure products.[2]
Furthermore,[7] the products of auxiliary-directed reactions are diastereomers, which enables their facile separation by methods such as column chromatography or crystallization.
8-phenylmenthol
In an early example of the use of a chiral auxiliary in asymmetric synthesis, E. J. Corey and coworkers conducted an asymmetric Diels-Alder reaction between (−)-8-phenylmenthol acrylateester and 5-benzyloxymethylcyclopentadiene.[5] The cycloaddition product was carried forward to the iodolactone shown below, an intermediate in the classic Corey synthesis of the prostaglandins. It is proposed that the back face of the acrylate is blocked by the auxiliary, so that cycloaddition occurs at the front face of the alkene.
Diastereoselective Diels-Alder cycloaddition with the chiral auxiliary (−)-8-phenylmenthol in route to the prostaglandins
(−)-8-phenylmenthol can be prepared from either enantiomer of pulegone,[8] though neither route is very efficient. Because of the widespread utility of the 8-phenylmenthol auxiliary, alternative compounds that are more easily synthesized, such as trans-2-phenyl-1-cyclohexanol[9] and trans-2-(1-pheyl-1-methylethyl)cyclohexanol[10] have been explored.
Hisashi Yamamoto first utilized (R)-BINOL as a chiral auxiliary in the asymmetric synthesis of limonene, which is an example of cyclic mono-terpenes. (R)-BINOL mononeryl ether was prepared by the monosilylation and alkylation of (R)-BINOL as the chiral auxiliary. Followed with the reduction by organoaluminum reagent, limonene was synthesized with low yields (29% yield) and moderate enantiomeric excesses up to 64% ee.[12]
First utilization of BINOL as a chiral auxiliary
The preparation of a variety of enantiomerically pure uncommon R-amino acids can be achieved by the alkylation of chiral glycine derivatives possessing axially chiral BINOL as an auxiliary. It has been depicted by Fuji et al. Based on different electrophile, the diastereomeric excess varied from 69% to 86.[13]
Diastereoselective addition between Grignard and BINOL protected aldehyde
Protected at the aldehyde function with (R)-BINOL, arylglyoxals reacted diastereoselectively with Grignard reagents to afford protected atrolactaldehyde with moderate to excellent diastereomeric excess and high yields.[14]
Diastereoselective addition between Grignard and BINOL protected aldehyde
BINOL was also used as a chiral auxiliary to control the formation of a P-stereocenter in an asymmetric metal-catalyzed C-P coupling process. Mondal et al. discovered that the Pd-catalysed C-P cross-coupling reaction between axially chiral BINOL-based phosphoramidites and aryl halides or triflates proceeds with excellent stereoselectivity due to the presence of BINOL near the reacting P center.[15]
trans-2-Phenylcyclohexanol was used in ene reaction as a chiral auxiliary.
In the total synthesis of (−)-heptemerone B and (−)-guanacastepene E, attached with trans-2-phenylcyclohexanol, the glyoxylate reacted with 2,4-dimethyl-pent-2-ene, in the presence of tin(IV) chloride, yielding the desired anti adduct as the major product, together with a small amount of its syn isomer with 10:1 diastereomeric ratio.[17]
The glyoxylate reacted with 2,4-dimethyl-2-pentane with trans-2-phenylcyclohexanol as a chiral auxiliary
For even greater conformational control, switching from a phenyl to a trityl group gives trans-2-tritylcyclohexanol (TTC). In 2015, the Brown group published an efficient chiral permanganate-mediated oxidative cyclization with TTC.[18]
trans-2-Tritylcyclohexanol was used in asymmetric permanganate-mediated oxidative cyclization.
Oxazolidinones
Oxazolidinone auxiliaries, popularized by David A. Evans, have been applied to many stereoselective transformations, including aldol reactions,[19]alkylation reactions,[20] and Diels-Alder reactions.[21][22] The oxazolidinones are substituted at the 4 and 5 positions. Through steric hindrance, the substituents direct the direction of substitution of various groups. The auxiliary is subsequently removed e.g. through hydrolysis.
Preparation
Oxazolidinones can be prepared from amino acids or readily available amino alcohols. A large number of oxazolidinones are commercially available, including the four below.
Evans auxiliaries
Some of the commercially available oxazolidinone chiral auxiliaries
Chiral oxazolidinones have been employed most widely in stereoselective aldol reactions.
Soft enolization with the Lewis acid dibutylboron triflate and the base diisopropylethylamine gives the (Z)-enolate, which undergoes a diastereoselective aldol reaction with an aldehyde substrate. The transformation is particularly powerful because it establishes two contiguous stereocenters simultaneously.
Stereoselective Evans aldol reaction
A model for the observed stereoselectivity can be found below. The syn-stereo relationship between the methyl group and the new secondary alcohol results from a six-membered ring Zimmerman-Traxler transition state, wherein the enolate oxygen and the aldehyde oxygen both coordinate boron. The aldehyde is oriented such that the hydrogen is placed in a pseudo-axial orientation to minimize 1,3-diaxial interactions. The absolute stereochemistry of the two stereocenters is controlled by the chirality in the auxiliary. In the transition structure, the auxiliary carbonyl is oriented away from the enolate oxygen so as to minimize the net dipole of the molecule; one face of the enolate is blocked by the substituent on the chiral auxiliary.
Model for the stereoselectivity of an Evans aldol reaction
Removal
A variety of transformations have been developed to facilitate removal of the oxazolidinone auxiliary to generate different synthetically useful functional groups.
Transformation of an oxazolidinone imide to different functional groups
Camphorsultam
Camphorsultam, or Oppolzer's sultam, is a classic chiral auxiliary.
Camphorsultam
In the total synthesis of manzacidin B, Ohfune group utilized camphorsultam to construct the core oxazoline ring asymmetrically. Comparing with oxazolidinone as the chiral auxiliary, camphorsultam had a significant (2S,3R)-selectivity.[23]
The chiral camphorsultam was found to be a superior chiral auxiliary to the oxazolidinone in view of the single asymmetric induction.
Camphorsultam also acts as a chiral auxiliary in Michael addition. Lithium base promoted stereoselective Michael addition of thiols to N-mcthacryloylcamphorsultam produced the corresponding addition products in high diastereoselectivity.[24]
Camphorsultam was used as a chiral auxiliary in Michael addition.
Camphorsultam was used as a chiral auxiliary for the asymmetric Claisen rearrangement. In the presence of butylated hydroxytoluene (BHT) used as a radical scavenger, a toluene solution of the adduct between geraniol and camphorsultam was heated in a sealed tube at 140°C, to provide mainly the (2R,3S)-isomer as the major rearrangement product in 72% yield, securing the two contiguous stereocenters including the quaternary carbon.[25]
Camphorsultam was used as a chiral auxiliary in Claisen rearrangement.
The α-proton of the carbonyl compound is easily deprotonated by a non-nucleophilic base to give the enolate, which can further react. The configuration of the addition compound, such as with an alkyl halide, is directed by the methyl group. Thus, any addition product will be syn with the methyl and anti to the hydroxyl group. The pseudoephedrine chiral auxiliary is subsequently removed by cleaving the amide bond with an appropriate nucleophile.
Preparation
Both enantiomers of pseudoephedrine are commercially available. Racemic pseudoephedrine has many medical uses. Because pseudoephedrine can be used to illegally make methamphetamine, the purchase of pseudoephedrine for use in academic or industrial research is rather regulated. As an alternative, Myers et al. reported the utility of pseudoephenamine chiral auxiliaries in alkylation reactions.[27] While pseudoephenamine is not readily available from commercial sources, it can be synthesized with relative ease from benzil and cannot be used to make amphetamines.[28]
Pseudoephedrine and pseudoephenamine chiral auxiliaries
Pseudoephedrine amides are typically prepared by acylation with an acyl chloride or anhydride.[29]
Acylation of (S,S)-psueodphedrine with an acid anhydride
Alkylation
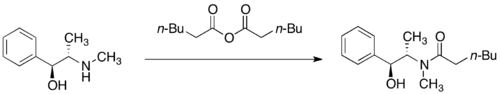
Pseudoephedrine amides undergo deprotonation by a strong base such as lithium diisopropylamide (LDA) to give the corresponding (Z)-enolates. Alkylation of these lithium enolates proceeds with high facial selectivity.
Diastereoselective alkylation of a pseudoephedrine amide
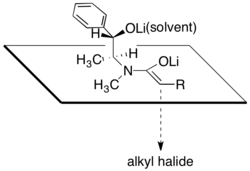
The diastereoselectivity is believed to result from a configuration wherein one face of the lithium enolate is blocked by the secondary lithium alkoxide and the solvent molecules associated with that lithium cation. In accordance with this proposal, it has been observed that the diastereoselectivity of the alkylation step is highly dependent on the amount of lithium chloride present and on the solvent, tetrahydrofuran (THF). Typically, 4 to 6 equivalents of lithium chloride are sufficient to saturate a solution of enolate in THF at the reaction molarity.
Model for the diastereoslective alkylation of a pseudoephedrine amide enolate
One primary advantage of asymmetric alkylation with pseudoephedrine amides is that the amide enolates are typically nucleophilic enough to react with primary and even secondary halides at temperatures ranging from –78°C to 0°C. Construction of quaternary carbon centers by alkylation of α-branched amide enolates is also possible, though the addition of DMPU is necessary for less reactive electrophiles.[30]
Removal
Conditions have been developed for the transformation of pseudoephedrine amides into enantiomerically enriched carboxylic acids, alcohols, aldehydes, and ketones - after cleavage, the auxiliary can be recovered and reused.
Transformation of pseudoephedrine amides into synthetically useful functional groups
This specific sulfinamide chiral auxiliary was initially developed by Jonathan A. Ellman, and its use has been explored extensively by his group.[31][32] Thus, it is often referred to as Ellman's auxiliary or Ellman's sulfinamide.
The two possible enantiomers of tert-butanesulfinamide
Preparation
Either enantiomer of tert-butanesulfinamide can be reached from tert-butyl disulfide in two steps: a catalytic asymmetric oxidation reaction gives the disulfide oxidation product (thiosulfinate) in high yield and enantiomeric excess. Treatment of this compound with lithium amide in ammonia affords optically pure inverted product.
Synthesis of tert-butanesulfinamide from readily available chemicals
Condensation of tert-butanesulfinamide with an aldehyde or ketone proceeds in high yield and affords only the (E)-isomer of the corresponding N-sulfinyl imines.
Condensation of tert-butanesulfinamide with aldehydes and ketones
Synthesis of chiral amines
Addition of a Grignard reagent to a tert-butanesulfinyl aldimine or ketimine results in asymmetric addition to give the branched sulfinamide. The observed stereoselectivity can be rationalized by a six-membered ring transition structure, wherein both oxygen and nitrogen of the sulfinyl imine coordinate magnesium.
Addition of a Grignard reagent to a tert-butanesulfinyl aldimine
Alkylation reactions of chiral (S)-1-amino-2-methoxymethylpyrrolidine (SAMP) and (R)-1-amino-2-methoxymethylpyrrolidine (RAMP) hydrazones were developed by Dieter Enders and E.J. Corey.[33][34]
Preparation
SAMP can be prepared in six steps from (S)-proline, and RAMP can be prepared in six steps from (R)-glutamic acid.
Preparation of SAMP and RAMP from commercially available materials.
Alkylation reactions
Condensation of SAMP or RAMP with an aldehyde or ketone affords the (E)-hydrazine. Deprotonation with lithium diisopropylamide and addition of an alkyl halide affords the alkylated product. The auxiliary can be removed by ozonolysis or hydrolysis.
Condensation, alkylation, and cleavage with SAMP/RAMP chiral auxiliaries
Chiral auxiliaries in industry
Chiral auxiliaries are generally reliable and versatile, enabling the synthesis of a large number of enantiomerically pure compounds in a time-efficient manner. Consequently, chiral auxiliaries are often the method of choice in the early phases of drug development.[2]
Tipranavir
The HIV protease inhibitor Tipranavir is marketed for the treatment of AIDS. The first enantioselective medicinal chemistry route to Tipranavir included the conjugate addition of an organocuprate reagent to a chiral Michael acceptor.[35] The chiral oxazolidinone in the Michael acceptor controlled the stereochemistry of one of two stereocenters in the molecule. The final, commercial route to Tipranavir does not feature a chiral auxiliary; instead, this stereocenter is set by an asymmetric hydrogenation reaction.[36]
Synthetic strategies for setting a key stereocenter in Tipranavir
Atorvastatin
The calcium salt of atorvastatin is marketed under the trade name Lipitor for the lowering of blood cholesterol. The first enantioselective medicinal chemistry route to atorvastatin relied on a diastereoselective aldol reaction with a chiral ester to set one of the two alcohol stereocenters.[37] In the commercial route to atorvastatin, this stereocenter is carried forward from the readily available food additiveisoascorbic acid.[38]
Synthetic strategies for setting a key stereocenter in atorvastatin
12Evans, D. A.; Helmchen, G.; Rüping, M. (2007). "Chiral Auxiliaries in Asymmetric Synthesis". In Christmann, M (ed.). Asymmetric Synthesis — The Essentials. Wiley-VCH Verlag GmbH & Co. pp.3–9. ISBN978-3-527-31399-0.
↑Nicolau, K. C. (2008). Classics in Total Synthesis (5thed.). New York, New York: Wiley-VCH. pp.485–508. ISBN978-3-527-29231-8.
↑Miller, J. P. (2013). "ChemInform Abstract: Recent Advances in Asymmetric Diels-Alder Reactions". ChemInform. 44 (48): no. doi:10.1002/chin.201348243.
↑Corey, E. J.; Ensley, H. E.; Parnell, C. A. (1978). "Convenient Synthesis of a Highly Efficient and Recyclable Chiral Director for Asymmetric Induction". J. Org. Chem.43 (8): 1610–1611. doi:10.1021/jo00402a037.
↑Whitesell, J. K.; Chen, H. H.; Lawrence, R. M. (1985). "trans-2-Phenylcyclohexanol. A powerful and readily available chiral auxiliary". J. Org. Chem. 50 (23): 4663–4664. doi:10.1021/jo00223a055.
↑Comins, D. L.; Salvador, J. D. (1993). "Efficient Synthesis and Resolution of trans-2-(1-Aryl-1-methylethyl)cyclohexanols: Practical Alternatives to 8-P henylmenthol". J. Org. Chem. 58 (17): 4656–4661. doi:10.1021/jo00069a031.
↑Brunel, Jean Michel (2005). "BINOL: A Versatile Chiral Reagent". Chemical Reviews. 105 (3): 857–898. doi:10.1021/cr040079g. PMID15755079.
12Sakane, Soichi; Fujiwara, Junya; Maruoka, Keiji; Yamamoto, Hisashi (1983). "Chiral leaving group. Biogenetic-type asymmetric synthesis of limonene and bisabolenes". Journal of the American Chemical Society. 105 (19): 6154–6155. Bibcode:1983JAChS.105.6154S. doi:10.1021/ja00357a033.
↑Tanaka, Kiyoshi; Ahn, Mija; Watanabe, Yukari; Fuji, Kaoru (1996-06-01). "Asymmetric synthesis of uncommon α-amino acids by diastereoselective alkylations of a chiral glycine equivalent". Tetrahedron: Asymmetry. 7 (6): 1771–1782. doi:10.1016/0957-4166(96)00212-1.
↑Maglioli, Paola; De Lucchi, Ottorino; Delogu, Giovanna; Valle, Giovanni (1992-01-01). "Highly diastereoselective reduction and addition of nucleophiles to binaphthol-protected arylglyoxals". Tetrahedron: Asymmetry. 3 (3): 365–366. doi:10.1016/S0957-4166(00)80276-1.
↑Buchi, George; Vogel, Dennis E. (1985). "A new method for the preparation of γ,δ-unsaturated ketones via Claisen rearrangement". The Journal of Organic Chemistry. 50 (23): 4664–4665. doi:10.1021/jo00223a056.
↑Miller, Aubry K.; Hughes, Chambers C.; Kennedy-Smith, Joshua J.; Gradl, Stefan N.; Dirk Trauner (2006). "Total Synthesis of (−)-Heptemerone B and (−)-Guanacastepene E". Journal of the American Chemical Society. 128 (51): 17057–17062. Bibcode:2006JAChS.12817057M. doi:10.1021/ja0660507. PMID17177458.
↑Al Hazmi, Ali M.; Sheikh, Nadeem S.; Bataille, Carole J. R.; Al-Hadedi, Azzam A. M.; Watkin, Sam V.; Luker, Tim J.; Camp, Nicholas P.; Brown, Richard C. D. (2014). "trans-2-Tritylcyclohexanol as a Chiral Auxiliary in Permanganate-Mediated Oxidative Cyclization of 2-Methylenehept-5-enoates: Application to the Synthesis of trans-(+)-Linalool Oxide". Organic Letters. 16 (19): 5104–5107. doi:10.1021/ol502454r. PMID25225741.
↑Evans, D. A.; Bartroli, J.; Shih, T. L. (1981). "Enantioselective aldol condensations. 2. Erythro-selective chiral aldol condensations via boron enolates". J. Am. Chem. Soc. 103 (8): 2127–2129. Bibcode:1981JAChS.103.2127E. doi:10.1021/ja00398a058.
↑Evans, D. A.; Ennis, M. D.; Mathre, D. J. (1982). "Asymmetric Alkylation Reactions of Chiral Imide Enolates. A Practical Approach to the Enantioselective Synthesis of a-Substituted Carboxylic Acid Derivatives". J. Am. Chem. Soc. 104 (6): 1737–1739. doi:10.1021/ja00370a050.
↑Evans, D. A.; Chapman, K. T.; Bisaha, J. (1984). "New Asymmetric Diels-Alder Cycloaddition Reactions. Chiral α,β-Unsaturated Carboximides as Practical Chiral Acrylate and Crotonate Dienophile Synthons". J. Am. Chem. Soc. 106 (15): 4261–4263. Bibcode:1984JAChS.106.4261E. doi:10.1021/ja00327a031.
↑Evans, D. A.; Chapman, K. T.; Hung, D. T.; Kawaguchi, A. T. (1987). "Transition State π-Solvation by Aromatic Rings: An Electronic Contribution to Diels-Alder Reaction Diastereoselectivity". Angew. Chem. Int. Ed.26 (11): 1184–1186. doi:10.1002/anie.198711841.
↑Shinada, Tetsuro; Oe, Kentaro; Ohfune, Yasufumi (2012-06-27). "Efficient total synthesis of manzacidin B". Tetrahedron Letters. 53 (26): 3250–3253. doi:10.1016/j.tetlet.2012.04.042.
↑Tsai, Wen-Jiuan; Lin, Yi-Tsong; Uang, Biing-Jiun (1994-07-01). "Asymmetric Michael addition of thiols to (1R,2R,4R)-(−)-2,10-N-enoylcamphorsultam". Tetrahedron: Asymmetry. 5 (7): 1195–1198. doi:10.1016/0957-4166(94)80155-X.
↑Myers, A. G.; etal. (1997). "Pseudoephedrine as a Practical Chiral Auxiliary for the Synthesis of Highly Enantiomerically Enriched Carboxylic Acids, Alcohols, Aldehydes, and Ketones". J. Am. Chem. Soc. 119 (28): 6496–6511. Bibcode:1997JAChS.119.6496M. doi:10.1021/ja970402f.
↑Myers, A. G.; Yang, B. H.; McKinstry, L.; Kopecky, D. J.; Gleason, J. L. (1997). "Pseudoephedrine as a Practical Chiral Auxiliary for the Synthesis of Highly Enantiomerically Enriched Carboxylic Acids, Alcohols, Aldehydes, and Ketones". J. Am. Chem. Soc. 119 (28): 6496–6511. Bibcode:1997JAChS.119.6496M. doi:10.1021/ja970402f.
↑Liu, Guangcheng; Cogan, Derek A.; Ellman, Jonathan A. (October 1997). "Catalytic Asymmetric Synthesis of tert -Butanesulfinamide. Application to the Asymmetric Synthesis of Amines". Journal of the American Chemical Society. 119 (41): 9913–9914. Bibcode:1997JAChS.119.9913L. doi:10.1021/ja972012z. ISSN0002-7863.
↑Ellman, J. A.; Owens, T. D.; Tang, T. P. (2002). "N-tert-Butanesulfinyl Imines: Versatile Intermediates for the Asymmetric Synthesis of Amines". Acc. Chem. Res.35 (11): 984–995. doi:10.1021/ar020066u. PMID12437323.
↑Corey, E. J.; Enders, D. (1976). "Applications of N,N-dimethylhydrazones to synthesis. Use in efficient, positionally and stereochemically selective C-C bond formation; oxidative hydrolysis to carbonyl compounds". Tetrahedron Letters. 17 (1): 3–6. doi:10.1016/s0040-4039(00)71307-4.
↑Kurti, L.; Czako, B. (2005). Strategic Applications of Named Reactions in Organic Synthesis. Burlington, MA: Elsevier Academic Press. pp.150–151. ISBN978-0-12-369483-6.
↑Turner, S. T.; etal. (1998). "Tipranavir (PNU-140690): A Potent, Orally Bioavailable Nonpeptidic HIV Protease Inhibitor of the 5,6-Dihydro-4-hydroxy-2-pyrone Sulfonamide Class". J. Med. Chem.41 (18): 3467–3476. doi:10.1021/jm9802158. PMID9719600.
↑Roth, B. D.; etal. (1991). "Inhibitors of Cholesterol Biosynthesis. 3. Tetrahydro-4-hydroxy-6-[2-(lH-pyrrol-l-yl)ethyl]-2H-pyran-2-one Inhibitors of HMG-CoA Reductase. 2. Effects of Introducing Substituents at Positions Three and Four of the Pyrrole Nucleus". J. Med. Chem. 34 (1): 357–366. doi:10.1021/jm00105a056. PMID1992137.
↑Jie Jack Li; Douglas S. Johnson; Drago R. Sliskovic; Bruce D. Roth (2004). "Chapter 9. Atorvastatin Calcium (Lipitor)". Contemporary Drug Synthesis. John Wiley & Sons, Inc. pp.113–125. ISBN978-0-471-21480-9.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.