Asymmetric hydrogenation is a chemical reaction that adds two atoms of hydrogen to a target (substrate) molecule with three-dimensional spatial selectivity. Critically, this selectivity does not come from the target molecule itself, but from other reagents or catalysts present in the reaction. This allows spatial information (what chemists refer to as chirality) to transfer from one molecule to the target, forming the product as a single enantiomer. The chiral information is most commonly contained in a catalyst and, in this case, the information in a single molecule of catalyst may be transferred to many substrate molecules, amplifying the amount of chiral information present. Similar processes occur in nature, where a chiral molecule like an enzyme can catalyse the introduction of a chiral centre to give a product as a single enantiomer, such as amino acids, that a cell needs to function. By imitating this process, chemists can generate many novel synthetic molecules that interact with biological systems in specific ways, leading to new pharmaceutical agents and agrochemicals. The importance of asymmetric hydrogenation in both academia and industry contributed to two of its pioneers — William Standish Knowles and Ryōji Noyori — being collectively awarded one half of the 2001 Nobel Prize in Chemistry.[1]
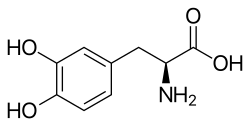
In 1956 a heterogeneous catalyst made of palladium deposited on silk was shown to effect asymmetric hydrogenation.[2] Later, in 1968, the groups of William Knowles and Leopold Horner independently published the examples of asymmetric hydrogenation using a homogeneous catalysts. While exhibiting only modest enantiomeric excesses, these early reactions demonstrated feasibility. By 1972, enantiomeric excess of 90% was achieved, and the first industrial synthesis of the Parkinson's drug L-DOPA commenced using this technology.[3][4]
L-DOPA
The field of asymmetric hydrogenation continued to experience a number of notable advances. Henri Kagan developed DIOP, an easily prepared C2-symmetric diphosphine that gave high ee's in certain reactions. Ryōji Noyori introduced the ruthenium-based catalysts for the asymmetric hydrogenated polar substrates, such as ketones and aldehydes. Robert H. Crabtree demonstrated the ability for Iridium compounds to catalyse asymmetric hydrogenation reactions in 1979 with the invention of Crabtree's catalyst.[5] In the early 1990's, the introduction of P,N ligands by several groups independently then further expanded the scope of the C2-symmetric ligands, although they are not fundamentally superior to chiral ligands lacking rotational symmetry.[6]
Today, asymmetric hydrogenation is a routine methodology in laboratory and industrial scale organic chemistry. The importance of asymmetric hydrogenation was recognized by the 2001 Nobel Prize in Chemistry awarded to William Standish Knowles and Ryōji Noyori.
Mechanism
Asymmetric hydrogenations operate by conventional mechanisms invoked for other hydrogenations. This includes inner sphere mechanisms, outer sphere mechanisms and the σ-bond metathesis mechanisms.[7] The type of mechanism employed by a catalyst is largely dependent on the ligands used in a system, which in turn leads to certain catalyst-substrate affinities.
Inner sphere mechanisms
The so-called inner sphere mechanism entails coordination of the alkene to the metal center.[8] Other characteristics of this mechanism include a tendency for a homolytic splitting of dihydrogen when more electron-rich, low-valent metals are present while electron-poor, high valent metals normally exhibit a heterolytic cleavage of dihydrogen assisted by a base.[9]
The diagram below depicts purposed mechanisms for catalytic hydrogenation with rhodium complexes which are inner sphere mechanisms. In the unsaturated mechanism, the chiral product formed will have the opposite mode compared to the catalyst used. While the thermodynamically favoured complex between the catalyst and the substrate is unable to undergo hydrogenation, the unstable, unfavoured complex undergoes hydrogenation rapidly.[10] The dihydride mechanism on the other hand sees the complex initially hydrogenated to the dihydride form. This subsequently allows for the coordination of the double bond on the non-hindered side. Through insertion and reductive elimination, the product's chirality matches that of the ligand.[11]
Proposed mechanisms for asymmetric hydrogenation
The preference for producing one enantiomer instead of another in these reactions is often explained in terms of steric interactions between the ligand and the prochiral substrate. Consideration of these interactions has led to the development of quadrant diagrams where "blocked" areas are denoted with a shaded box, while "open" areas are left unfilled. In the modeled reaction, large groups on an incoming olefin will tend to orient to fill the open areas of the diagram, while smaller groups will be directed to the blocked areas and hydrogen delivery will then occur to the back face of the olefin, fixing the stereochemistry. Note that only part of the chiral phosphine ligand is shown for the sake of clarity.
Quadrant model for asymmetric hydrogenation
Outer sphere mechanisms
Some catalysts operate by "outer sphere mechanisms" such that the substrate never bonds directly to the metal but rather interacts with its ligands, which is often a metal hydride and a protic hydrogen on a ligand. As such, in most cases dihydrogen is split heterolytically, with the metal acting as a Lewis acid and either an external or internal base "deprotonating" the hydride.[7]
Proposed intermediates in the outer sphere mechanisms for: heterolytic hydrogenation of the catalyst (left) and hydride transfer from the catalyst to the substrate (right)
For an example of this mechanism we can consider the BINAP-Ru-diamine system. The dihalide form of the catalyst is converted to the catalysts by reaction of H2 in the presence of base:[12]
The "Noyori-class" of catalysts are often referred to as bifunctional catalysts to emphasize the fact that both the metal and the (amine) ligand are functional.[13]
In the hydrogenation of C=O containing substrates, the mechanism was long assumed to operate by a six membered pericyclic transition state/intermediate whereby the hydrido ruthenium hydride center (HRu-NH) interacts with the carbonyl substrate R2C=O.[14] More recent DFT and experimental studies have shown that this model is largely incorrect. Instead, the amine backbone interacts strongly with the base activator, which often is used in large excess.[12] However in both cases, the substrate does not bond directly with the metal centre, thus making it a great example of an outer sphere mechanism.
Iron is a popular research target for many catalytic processes, owing largely to its low cost and low toxicity relative to other transition metals.[18] Asymmetric hydrogenation methods using iron have been realized, although in terms of rates and selectivity, they are inferior to catalysts based on precious metals.[19] In some cases, structurally ill-defined nanoparticles have proven to be the active species in situ and the modest selectivity observed may result from their uncontrolled geometries.[20]
Chiral phosphine ligands can be generally classified as mono- or bidentate. They can be further classified according to the location of the stereogenic centre – phosphorus vs the organic substituents. Ligands with a C2 symmetry element have been particularly popular, in part because the presence of such an element reduces the possible binding conformations of a substrate to a metal-ligand complex dramatically (often resulting in exceptional enantioselectivity).[21]
Monodentate phosphines
Monophosphine-type ligands were among the first to appear in asymmetric hydrogenation, e.g., the ligand CAMP.[22] Continued research into these types of ligands has explored both P-alkyl and P-heteroatom bonded ligands, with P-heteroatom ligands like the phosphites and phosphoramidites generally achieving more impressive results.[23] Structural classes of ligands that have been successful include those based on the binapthyl structure of MonoPHOS [24] or the spiro ring system of SiPHOS.[25] Notably, these monodentate ligands can be used in combination with each other to achieve a synergistic improvement in enantioselectivity;[26] something that is not possible with the diphosphine ligands.[23]
The diphosphine ligands have received considerably more attention than the monophosphines and, perhaps as a consequence, have a much longer list of achievement. This class includes the first ligand to achieve high selectivity (DIOP), the first ligand to be used in industrial asymmetric synthesis (DIPAMP[27][28][4]) and what is likely the best known chiral ligand (BINAP).[3] Chiral diphosphine ligands are now ubiquitous in asymmetric hydrogenation.
Historically important diphosphine ligands
P,N and P,O ligands
Generic PHOX ligand architecture
Effective ligand for various asymmetric-hydrogenation processes
The use of P,N ligands in asymmetric hydrogenation can be traced to the C2 symmetric bisoxazoline ligand.[29] However, these symmetric ligands were soon superseded by monooxazoline ligands whose lack of C2 symmetry has in no way limits their efficacy in asymmetric catalysis.[30] Such ligands generally consist of an achiral nitrogen-containing heterocycle that is functionalized with a pendant phosphorus-containing arm, although both the exact nature of the heterocycle and the chemical environment phosphorus center has varied widely. No single structure has emerged as consistently effective with a broad range of substrates, although certain privileged structures (like the phosphine-oxazoline or PHOX architecture) have been established.[31][30][32] Moreover, within a narrowly defined substrate class the performance of metallic complexes with chiral P,N ligands can closely approach perfect conversion and selectivity in systems otherwise very difficult to target.[33] Certain complexes derived from chelating P-O ligands have shown promising results in the hydrogenation of α,β-unsaturated ketones and esters.[34]
NHC ligands
Catalyst developed by Burgess for asymmetric hydrogenation
Simple N-heterocyclic carbene (NHC)-based ligands have proven impractical for asymmetrical hydrogenation.
Some C,N ligands combine an NHC with a chiral oxazoline to give a chelating ligand.[35][36] NHC-based ligands of the first type have been generated as large libraries from the reaction of smaller libraries of individual NHCs and oxazolines.[35][36] NHC-based catalysts featuring a bulky seven-membered metallocycle on iridium have been applied to the catalytic hydrogenation of unfunctionalized olefins[35] and vinyl ether alcohols with conversions and ee's in the high 80s or 90s.[37] The same system has been applied to the synthesis of a number of aldol,[38] vicinal dimethyl[39] and deoxypolyketide[40] motifs, and to the deoxypolyketides themselves.[41]
C2-symmetric NHCs have shown themselves to be highly useful ligands for the asymmetric hydrogenation.[42]
Acyclic substrates
Substrates can be classified according to their polarity. Nonpolar substrates are dominated by alkenes. Polar substrates include ketones, enaminesketimines.
Alkenes that are particularly amenable to asymmetric hydrogenation often feature a polar functional group adjacent to the site to be hydrogenated. In the absence of this functional group, catalysis often results in low ee's. For some unfunctionalized olefins, iridium with P,N-based ligands) have proven effective, however. Alkene substrates are often classified according to their substituents, e.g., 1,1-disubstituted, 1,2-diaryl trisubstituted, 1,1,2-trialkyl and tetrasubstituted olefins.[44][45] and even within these classes variations may exist that make different solutions optimal.[46]
Example of asymmetric hydrogenation of unfunctionalized olefins
Chiral phosphoramidite and phosphonite ligands used in the asymmetric hydrogenation of enamines
Conversely to the case of olefins, asymmetric hydrogenation of enamines has favoured diphosphine-type ligands; excellent results have been achieved with both iridium- and rhodium-based systems. However, even the best systems often suffer from low ee's and a lack of generality. Certain pyrrolidine-derived enamines of aromatic ketones are amenable to asymmetrically hydrogenation with cationic rhodium(I) phosphonite systems, and I2 and acetic acid system with ee values usually above 90% and potentially as high as 99.9%.[47] A similar system using iridium(I) and a very closely related phosphoramidite ligand is effective for the asymmetric hydrogenation of pyrrolidine-type enamines where the double bond was inside the ring: in other words, of dihydropyrroles.[48] In both cases, the enantioselectivity dropped substantially when the ring size was increased from five to six.
Imines and ketones
Noyori catalyst for asymmetric hydrogenation of ketones
Ketones and imines are related functional groups, and effective technologies for the asymmetric hydrogenation of each are also closely related. Early examples are Noyori's ruthenium-chiral diphosphine-diamine system.[49][50]
For carbonyl and imine substrates, end-on, η1 coordination can compete with η2 mode. For η1-bound substrates, the hydrogen-accepting carbon is removed from the catalyst and resists hydrogenation.[51]
Iridium/P,N ligand-based systems have been effective for some ketones and imines. For example, a consistent system for benzylic aryl imines uses the P,N ligand SIPHOX in conjunction with iridium(I) in a cationic complex to achieve asymmetric hydrogenation with ee >90%.[52] An efficient catalyst for ketones, (turnover number (TON) up to 4,550,000 and ee up to 99.9%) is an iridium(I) system with a closely related tridentate ligand.[53]
Highly effective system for the asymmetric hydrogenation of ketones
The BINAP/diamine-Ru catalyst is effective for the asymmetric reduction of both functionalized and simple ketones,[54] and BINAP/diamine-Ru catalyst can catalyze aromatic, heteroaromatic, and olefinicketones enantioselectively.[55] Better stereoselectivity is achieved when one substituent is larger than the other (see Flippin-Lodge angle).
BINAP/diamine-Ru catalyst scope
Aromatic substrates
The asymmetric hydrogenation of aromatic (especially heteroaromatic), substrates is a very active field of ongoing research. Catalysts in this field must contend with a number of complicating factors, including the tendency of highly stable aromatic compounds to resist hydrogenation, the potential coordinating (and therefore catalyst-poisoning) abilities of both substrate and product, and the great diversity in substitution patterns that may be present on any one aromatic ring.[56] Of these substrates the most consistent success has been seen with nitrogen-containing heterocycles, where the aromatic ring is often activated either by protonation or by further functionalization of the nitrogen (generally with an electron-withdrawing protecting group). Such strategies are less applicable to oxygen- and sulfur-containing heterocycles, since they are both less basic and less nucleophilic; this additional difficulty may help to explain why few effective methods exist for their asymmetric hydrogenation.
Quinolines, isoquinolines and quinoxalines
Two systems exist for the asymmetric hydrogenation of 2-substituted quinolines with isolated yields generally greater than 80% and ee values generally greater than 90%. The first is an iridium(I)/chiral phosphine/I2 system, first reported by Zhou et al..[57] While the first chiral phosphine used in this system was MeOBiPhep, newer iterations have focused on improving the performance of this ligand. To this end, systems use phosphines (or related ligands) with improved air stability,[58] recyclability,[58] ease of preparation,[59] lower catalyst loading[60][61] and the potential role of achiral phosphine additives.[62] As of October 2012 no mechanism appears to have been proposed, although both the necessity of I2 or a halogen surrogate and the possible role of the heteroaromatic N in assisting reactivity have been documented.[56]
The second is an organocatalytic transfer hydrogenation system based on Hantzsch esters and a chiral Brønsted acid. In this case, the authors envision a mechanism where the isoquinoline is alternately protonated in an activating step, then reduced by conjugate addition of hydride from the Hantzsch ester.[63]
Proposed organocatalytic mechanism
Much of the asymmetric hydrogenation chemistry of quinoxalines is closely related to that of the structurally similar quinolines. Effective (and efficient) results can be obtained with an Ir(I)/phophinite/I2 system[64] and a Hantzsh ester-based organocatalytic system,[65] both of which are similar to the systems discussed earlier with regards to quinolines.
Pyridines
Pyridines are highly variable substrates for asymmetric reduction (even compared to other heteroaromatics), in that five carbon centers are available for differential substitution on the initial ring. As of October 2012 no method seems to exist that can control all five, although at least one reasonably general method exists.
The most-general method of asymmetric pyridine hydrogenation is actually a heterogeneous method, where asymmetry is generated from a chiral oxazolidinone bound to the C2 position of the pyridine. Hydrogenating such functionalized pyridines over a number of different heterogeneous metal catalysts gave the corresponding piperidine with the substituents at C3, C4, and C5 positions in an all-cis geometry, in high yield and excellent enantioselectivity. The oxazolidinone auxiliary is also conveniently cleaved under acidic conditions as a second step.[66]
Asymmetric hydrogenation of pyridines with heterogeneous catalyst
Methods designed specifically for 2-substituted pyridine hydrogenation can involve asymmetric systems developed for related substrates like 2-substituted quinolines and quinoxalines. For example, an iridium(I)\chiral phosphine\I2 system is effective in the asymmetric hydrogenation of activated (alkylated) 2-pyridiniums[67] or certain cyclohexanone-fused pyridines.[68] Similarly, chiral Brønsted acid catalysis with a Hantzsh ester as a hydride source is effective for some 2-alkyl pyridines with additional activating substitution.[69]
The asymmetric hydrogenation of furans and benzofurans
Asymmetric hydrogenation of thiophenes and benzothiophenes has been catalyzed by some ruthenium(II) complexes of N-heterocyclic carbenes (NHC). This system appears to possess superb selectivity (ee > 90%) and perfect diastereoselectivity (all cis) if the substrate has a fused (or directly bound) phenyl ring but yields only racemic product in all other tested cases.[75]
The asymmetric hydrogenation of thiophenes and benzothiophenes
Heterogeneous catalysis
No heterogeneous catalyst has been commercialized for asymmetric hydrogenation.
The first asymmetric hydrogenation focused on palladium deposited on a silk support. Cinchonaalkaloids have been used as chiral modifiers for enantioselectivity hydrogenation.[76]
An alternative technique and one that allows more control over the structural and electronic properties of active catalytic sites is the immobilization of catalysts that have been developed for homogeneous catalysis on a heterogeneous support. Covalent bonding of the catalyst to a polymer or other solid support is perhaps most common, although immobilization of the catalyst may also be achieved by adsorption onto a surface, ion exchange, or even physical encapsulation. One drawback of this approach is the potential for the proximity of the support to change the behaviour of the catalyst, lowering the enantioselectivity of the reaction. To avoid this, the catalyst is often bound to the support by a long linker though cases are known where the proximity of the support can actually enhance the performance of the catalyst.[76]
The final approach involves the construction of MOFs that incorporate chiral reaction sites from a number of different components, potentially including chiral and achiral organic ligands, structural metal ions, catalytically active metal ions, and/or preassembled catalytically active organometallic cores.[77] One of these involved ruthenium-based catalysts. As little as 0.005mol% of such catalysts proved sufficient to achieve the asymmetric hydrogenation of aryl ketones, although the usual conditions featured 0.1mol% of catalyst and resulted in an enantiomeric excess of 90.6–99.2%.[78]
The active site of a heterogeneous zirconium phosphonate catalyst for asymmetric hydrogenation
Industrial applications
(S,S)-Ro 67-8867
Asymmetric hydrogenations are used in the production of several drugs, such as the antibacterial levofloxacin, the antibiotic carbapenem, and the antipsychotic agent BMS181100.[15][16][17]
Noyori asymmetric hydrogenation
Knowles' research into asymmetric hydrogenation and its application to the production scale synthesis of L-Dopa[4] gave asymmetric hydrogenation a strong start in the industrial world. A 2001 review indicated that asymmetric hydrogenation accounted for 50% of production scale, 90% of pilot scale, and 74% of bench scale catalytic, enantioselective processes in industry, with the caveat that asymmetric catalytic methods in general were not yet widely used.[79]
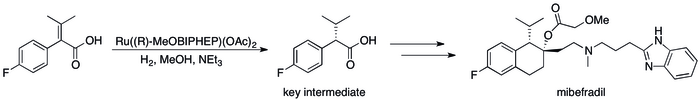
Asymmetric hydrogenation has replaced kinetic resolution based methods has resulted in substantial improvements in the process's efficiency.[12] can be seen in a number of specific cases where the For example, Roche's Catalysis Group was able to achieve the synthesis of (S,S)-Ro 67-8867 in 53% overall yield, a dramatic increase above the 3.5% that was achieved in the resolution based synthesis.[80] Roche's synthesis of mibefradil was likewise improved by replacing resolution with asymmetric hydrogenation, reducing the step count by three and increasing the yield of a key intermediate to 80% from the original 70%.[81]
Asymmetric hydrogenation in the industrial synthesis of mibefradil
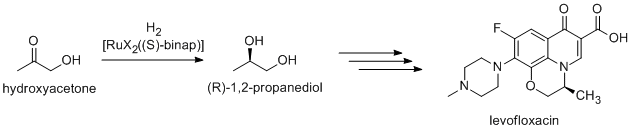
Noyori-inspired hydrogenation catalysts have been applied to the commercial synthesis of number of fine chemicals. (R)-1,2-Propandiol, precursor to the antibacterial levofloxacin, can be efficiently synthesized from hydroxyacetone using Noyori asymmetric hydrogenation:[17]
An antibiotic carbapenem is also prepared using Noyori asymmetric hydrogenation via (2S,3R)-methyl 2-(benzamidomethyl)-3-hydroxybutanoate, which is synthesized from racemic methyl 2-(benzamidomethyl)-3-oxobutanoate by dynamic kinetic resolution.
carbapenem synthesis
An antipsychotic agent BMS-181100 is synthesized using BINAP/diamine-Ru catalyst.
↑Gridnev, I. D.; Imamoto, T. (2004). "On the Mechanism of Stereoselection in Rh-Catalyzed Asymmetric Hydrogenation: A General Approach for Predicting the Sense of Enantioselectivity". Accounts of Chemical Research. 37 (9): 633–644. doi:10.1021/ar030156e. PMID15379579.
1234Dub, Pavel A.; Gordon, John C. (2018). "The role of the metal-bound N–H functionality in Noyori-type molecular catalysts". Nature Reviews Chemistry. 2 (12): 396–408. doi:10.1038/s41570-018-0049-z. S2CID106394152.
↑Noyori, Ryōji; Masashi Yamakawa; Shohei Hashiguchi (2001-11-01). "Metal−Ligand Bifunctional Catalysis: A Nonclassical Mechanism for Asymmetric Hydrogen Transfer between Alcohols and Carbonyl Compounds". The Journal of Organic Chemistry. 66 (24): 7931–7944. doi:10.1021/jo010721w. PMID11722188.
12Ikariya, T.; Blacker, A. J. (2007). "Asymmetric Transfer Hydrogenation of Ketones with Bifunctional Transition Metal-Based Molecular Catalysts". Accounts of Chemical Research. 40 (12): 1300–1308. doi:10.1021/ar700134q. PMID17960897.
12Pilkington, C.; Lennon, I. (2003). "The Application of Asymmetric Hydrogenation for the Manufacture of Pharmaceutical Intermediates:The Need for Catalyst Diversity". Synthesis. 2003 (11): 1639. doi:10.1055/s-2003-40871.
↑Enthaler, S.; Junge, K.; Beller, M. (2008). "Sustainable Metal Catalysis with Iron: From Rust to a Rising Star?". Angewandte Chemie International Edition. 47 (18): 3317–21. doi:10.1002/anie.200800012. PMID18412184.
↑Mikhailine, A.; Lough, A. J.; Morris, R. H. (2009). "Efficient Asymmetric Transfer Hydrogenation of Ketones Catalyzed by an Iron Complex Containing a P−N−N−P Tetradentate Ligand Formed by Template Synthesis". Journal of the American Chemical Society. 131 (4): 1394–1395. Bibcode:2009JAChS.131.1394M. doi:10.1021/ja809493h. PMID19133772.
↑Sonnenberg, J. F.; Coombs, N.; Dube, P. A.; Morris, R. H. (2012). "Iron Nanoparticles Catalyzing the Asymmetric Transfer Hydrogenation of Ketones". Journal of the American Chemical Society. 134 (13): 5893–5899. Bibcode:2012JAChS.134.5893S. doi:10.1021/ja211658t. PMID22448656.
↑Whitesell, J. K. (1989). "C2 symmetry and asymmetric induction". Chemical Reviews. 89 (7): 1581–1590. doi:10.1021/cr00097a012.
↑Knowles, W. S.; Sabacky, M. J.; Vineyard, B. D. (1972). "Catalytic asymmetric hydrogenation". Journal of the Chemical Society, Chemical Communications. 214 (1): 119–124. doi:10.1039/C39720000010. PMID4270504.
12Jerphagnon, T.; Renaud, J. L.; Bruneau, C. (2004). "Chiral monodentate phosphorus ligands for rhodium-catalyzed asymmetric hydrogenation". Tetrahedron: Asymmetry. 15 (14): 2101. doi:10.1016/j.tetasy.2004.04.037.
↑Vineyard, B. D.; Knowles, W. S.; Sabacky, M. J.; Bachman, G. L.; Weinkauff, D. J. (1977). "Asymmetric hydrogenation. Rhodium chiral bisphosphine catalyst". Journal of the American Chemical Society. 99 (18): 5946. Bibcode:1977JAChS..99.5946V. doi:10.1021/ja00460a018.
↑Knowles, W. S.; Sabacky, M. J.; Vineyard, B. D.; Weinkauff, D. J. (1975). "Asymmetric hydrogenation with a complex of rhodium and a chiral bisphosphine". Journal of the American Chemical Society. 97 (9): 2567. Bibcode:1975JAChS..97.2567K. doi:10.1021/ja00842a058.
↑Müller, D.; Umbricht, G.; Weber, B.; Pfaltz, A. (1991). "C2-Symmetric 4,4',5,5'-Tetrahydrobi(oxazoles) and 4,4',5,5'-Tetrahydro-2,2'-methylenebis[oxazoles] as Chiral Ligands for Enantioselective Catalysis Preliminary Communication". Helvetica Chimica Acta. 74: 232–240. doi:10.1002/hlca.19910740123.
12Helmchen, G. N.; Pfaltz, A. (2000). "PhosphinooxazolinesA New Class of Versatile, Modular P,N-Ligands for Asymmetric Catalysis". Accounts of Chemical Research. 33 (6): 336–345. doi:10.1021/ar9900865. PMID10891051.
↑Franzke, A.; Pfaltz, A. (2011). "Zwitterionic Iridium Complexes with P,N-Ligands as Catalysts for the Asymmetric Hydrogenation of Alkenes". Chemistry: A European Journal. 17 (15): 4131–44. doi:10.1002/chem.201003314. PMID21381140.
↑Maurer, F.; Huch, V.; Ullrich, A.; Kazmaier, U. (2012). "Development of Catalysts for the Stereoselective Hydrogenation of α,β-Unsaturated Ketones". The Journal of Organic Chemistry. 77 (11): 5139–5143. doi:10.1021/jo300246c. PMID22571628.
↑Rageot, D.; Woodmansee, D. H.; Pugin, B. T.; Pfaltz, A. (2011). "Proline-Based P,O Ligand/Iridium Complexes as Highly Selective Catalysts: Asymmetric Hydrogenation of Trisubstituted Alkenes". Angewandte Chemie International Edition. 50 (41): 9598–601. doi:10.1002/anie.201104105. PMID21882320.
123Perry, M. C.; Cui, X.; Powell, M. T.; Hou, D. R.; Reibenspies, J. H.; Burgess, K. (2003). "Optically Active Iridium Imidazol-2-ylidene-oxazoline Complexes: Preparation and Use in Asymmetric Hydrogenation of Arylalkenes". Journal of the American Chemical Society. 125 (1): 113–123. Bibcode:2003JAChS.125..113P. doi:10.1021/ja028142b. PMID12515512.
↑Zhu, Y.; Burgess, K. (2008). "Iridium-Catalyzed Asymmetric Hydrogenation of Vinyl Ethers". Advanced Synthesis & Catalysis. 350 (7–8): 979. doi:10.1002/adsc.200700546.
↑Zhao, J.; Burgess, K. (2009). "Aldol-Type Chirons from Asymmetric Hydrogenations of Trisubstituted Alkenes". Organic Letters. 11 (10): 2053–2056. doi:10.1021/ol900308w. PMID19368378.
↑Zhao, J.; Burgess, K. (2009). "Synthesis of Vicinal Dimethyl Chirons by Asymmetric Hydrogenation of Trisubstituted Alkenes". Journal of the American Chemical Society. 131 (37): 13236–13237. Bibcode:2009JAChS.13113236Z. doi:10.1021/ja905458n. PMID19719102.
↑Zhou, J.; Burgess, K. (2007). "Α,ω-Functionalized 2,4-Dimethylpentane Dyads and 2,4,6-Trimethylheptane Triads through Asymmetric Hydrogenation". Angewandte Chemie International Edition. 46 (7): 1129–31. doi:10.1002/anie.200603635. PMID17200966.
↑Zhou, J.; Zhu, Y.; Burgess, K. (2007). "Synthesis of (S,R,R,S,R,S)-4,6,8,10,16,18- Hexamethyldocosane from Antitrogus parvulus via Diastereoselective Hydrogenations". Organic Letters. 9 (7): 1391–1393. doi:10.1021/ol070298z. PMID17338543.
↑Urban, S.; Ortega, N.; Glorius, F. (2011). "Ligand-Controlled Highly Regioselective and Asymmetric Hydrogenation of Quinoxalines Catalyzed by Ruthenium N-Heterocyclic Carbene Complexes". Angewandte Chemie International Edition. 50 (16): 3803–6. doi:10.1002/anie.201100008. PMID21442699.
↑Cui, X.; Burgess, K. (2005). "Catalytic Homogeneous Asymmetric Hydrogenations of Largely Unfunctionalized Alkenes". Chemical Reviews. 105 (9): 3272–3296. doi:10.1021/cr0500131. PMID16159153.
↑Pàmies, O.; Andersson, P. G.; Diéguez, M. (2010). "Asymmetric Hydrogenation of Minimally Functionalised Terminal Olefins: An Alternative Sustainable and Direct Strategy for Preparing Enantioenriched Hydrocarbons". Chemistry: A European Journal. 16 (48): 14232–40. doi:10.1002/chem.201001909. PMID21140401.
↑Woodmansee, D. H.; Pfaltz, A. (2011). "Asymmetric hydrogenation of alkenes lacking coordinating groups". Chemical Communications. 47 (28): 7912–7916. doi:10.1039/c1cc11430a. PMID21556431.
↑Mazuela, J.; Verendel, J. J.; Coll, M.; SchäFfner, B. N.; BöRner, A.; Andersson, P. G.; PàMies, O.; DiéGuez, M. (2009). "Iridium Phosphite−Oxazoline Catalysts for the Highly Enantioselective Hydrogenation of Terminal Alkenes". Journal of the American Chemical Society. 131 (34): 12344–12353. Bibcode:2009JAChS.13112344M. doi:10.1021/ja904152r. PMID19658416.
↑Hou, G. H.; Xie, J. H.; Wang, L. X.; Zhou, Q. L. (2006). "Highly Efficient Rh(I)-Catalyzed Asymmetric Hydrogenation of Enamines Using Monodente Spiro Phosphonite Ligands". Journal of the American Chemical Society. 128 (36): 11774–11775. Bibcode:2006JAChS.12811774H. doi:10.1021/ja0644778. PMID16953614.
↑Hou, G. H.; Xie, J. H.; Yan, P. C.; Zhou, Q. L. (2009). "Iridium-Catalyzed Asymmetric Hydrogenation of Cyclic Enamines". Journal of the American Chemical Society. 131 (4): 1366–1367. Bibcode:2009JAChS.131.1366H. doi:10.1021/ja808358r. PMID19132836.
↑Hems, W. P.; Groarke, M.; Zanotti-Gerosa, A.; Grasa, G. A. (2007). "[(Bisphosphine) Ru(II) Diamine] Complexes in Asymmetric Hydrogenation: Expanding the Scope of the Diamine Ligand". Accounts of Chemical Research. 40 (12): 1340–1347. doi:10.1021/ar7000233. PMID17576143.
↑Noyori, R.; Yamakawa, M.; Hashiguchi, S. (2001). "Metal−Ligand Bifunctional Catalysis: A Nonclassical Mechanism for Asymmetric Hydrogen Transfer between Alcohols and Carbonyl Compounds". The Journal of Organic Chemistry. 66 (24): 7931–7944. doi:10.1021/jo010721w. PMID11722188.
↑Zhu, S. F.; Xie, J. B.; Zhang, Y. Z.; Li, S.; Zhou, Q. L. (2006). "Well-Defined Chiral Spiro Iridium/Phosphine−Oxazoline Cationic Complexes for Highly Enantioselective Hydrogenation of Imines at Ambient Pressure". Journal of the American Chemical Society. 128 (39): 12886–12891. Bibcode:2006JAChS.12812886Z. doi:10.1021/ja063444p. PMID17002383.
↑Xie, J. H.; Liu, X. Y.; Xie, J. B.; Wang, L. X.; Zhou, Q. L. (2011). "An Additional Coordination Group Leads to Extremely Efficient Chiral Iridium Catalysts for Asymmetric Hydrogenation of Ketones". Angewandte Chemie International Edition. 50 (32): 7329–32. doi:10.1002/anie.201102710. PMID21751315.
↑Ohkuma, T.; Ooka, H.; Yamakawa, M.; Ikariya, T.; Noyori, R. (1996), "Stereoselective Hydrogenation of Simple Ketones Catalyzed by Ruthenium(II) Complexes", The Journal of Organic Chemistry, 61 (15): 4872–4873, doi:10.1021/jo960997h
12Xu, L.; Lam, K. H.; Ji, J.; Wu, J.; Fan, Q. H.; Lo, W. H.; Chan, A. S. C. (2005). "Air-stable Ir-(P-Phos) complex for highly enantioselective hydrogenation of quinolines and their immobilization in poly(ethylene glycol) dimethyl ether (DMPEG)". Chemical Communications (11): 1390–2. doi:10.1039/B416397D. hdl:10397/8906. PMID15756313.
↑Lam, K. H.; Xu, L.; Feng, L.; Fan, Q. H.; Lam, F. L.; Lo, W. H.; Chan, A. S. C. (2005). "Highly Enantioselective Iridium-Catalyzed Hydrogenation of Quinoline Derivatives Using Chiral Phosphinite H8-BINAPO". Advanced Synthesis & Catalysis. 347 (14): 1755. doi:10.1002/adsc.200505130. hdl:10397/26878.
↑Wang, Z. J.; Deng, G. J.; Li, Y.; He, Y. M.; Tang, W. J.; Fan, Q. H. (2007). "Enantioselective Hydrogenation of Quinolines Catalyzed by Ir(BINAP)-Cored Dendrimers: Dramatic Enhancement of Catalytic Activity". Organic Letters. 9 (7): 1243–1246. doi:10.1021/ol0631410. PMID17328554.
↑Qiu, L.; Kwong, F. Y.; Wu, J.; Lam, W. H.; Chan, S.; Yu, W. Y.; Li, Y. M.; Guo, R.; Zhou, Z.; Chan, A. S. C. (2006). "A New Class of Versatile Chiral-Bridged Atropisomeric Diphosphine Ligands: Remarkably Efficient Ligand Syntheses and Their Applications in Highly Enantioselective Hydrogenation Reactions". Journal of the American Chemical Society. 128 (17): 5955–5965. Bibcode:2006JAChS.128.5955Q. doi:10.1021/ja0602694. hdl:10397/60397. PMID16637664.
↑Reetz, M. T.; Li, X. (2006). "Asymmetric hydrogenation of quinolines catalyzed by iridium complexes of BINOL-derived diphosphonites". Chemical Communications (20): 2159–60. doi:10.1039/b602320g. PMID16703140.
↑Rueping; Antonchick, A.; Theissmann, T. (2006). "A highly enantioselective Brønsted acid catalyzed cascade reaction: organocatalytic transfer hydrogenation of quinolines and their application in the synthesis of alkaloids". Angewandte Chemie International Edition in English. 45 (22): 3683–3686. doi:10.1002/anie.200600191. PMID16639754.
↑Tang, W.; Xu, L.; Fan, Q. H.; Wang, J.; Fan, B.; Zhou, Z.; Lam, K. H.; Chan, A. S. C. (2009). "Asymmetric Hydrogenation of Quinoxalines with Diphosphinite Ligands: A Practical Synthesis of Enantioenriched, Substituted Tetrahydroquinoxalines". Angewandte Chemie International Edition. 48 (48): 9135–8. doi:10.1002/anie.200904518. hdl:10397/20432. PMID19876991.
↑Rueping, M.; Tato, F.; Schoepke, F. R. (2010). "The First General, Efficient and Highly Enantioselective Reduction of Quinoxalines and Quinoxalinones". Chemistry: A European Journal. 16 (9): 2688–91. doi:10.1002/chem.200902907. PMID20140920.
↑Glorius, F.; Spielkamp, N.; Holle, S.; Goddard, R.; Lehmann, C. W. (2004). "Efficient Asymmetric Hydrogenation of Pyridines". Angewandte Chemie International Edition. 43 (21): 2850–2. doi:10.1002/anie.200453942. PMID15150766.
↑Ye, Z. S.; Chen, M. W.; Chen, Q. A.; Shi, L.; Duan, Y.; Zhou, Y. G. (2012). "Iridium-Catalyzed Asymmetric Hydrogenation of Pyridinium Salts". Angewandte Chemie International Edition. 51 (40): 10181–4. doi:10.1002/anie.201205187. PMID22969060.
↑Tang, W. J.; Tan, J.; Xu, L. J.; Lam, K. H.; Fan, Q. H.; Chan, A. S. C. (2010). "Highly Enantioselective Hydrogenation of Quinoline and Pyridine Derivatives with Iridium-(P-Phos) Catalyst". Advanced Synthesis & Catalysis. 352 (6): 1055. doi:10.1002/adsc.200900870. hdl:10397/22884.
↑Rueping, M.; Antonchick, A. P. (2007). "Organocatalytic Enantioselective Reduction of Pyridines". Angewandte Chemie International Edition. 46 (24): 4562–5. doi:10.1002/anie.200701158. PMID17492817.
↑Baeza, A.; Pfaltz, A. (2010). "Iridium-Catalyzed Asymmetric Hydrogenation of N-Protected Indoles". Chemistry: A European Journal. 16 (7): 2036–9. doi:10.1002/chem.200903105. PMID20104554.
↑Xiao, Y. C.; Wang, C.; Yao, Y.; Sun, J.; Chen, Y. C. (2011). "Direct Asymmetric Hydrosilylation of Indoles: Combined Lewis Base and Brønsted Acid Activation". Angewandte Chemie International Edition. 50 (45): 10661–4. doi:10.1002/anie.201105341. PMID21932274.
↑Duan, Y.; Chen, M. W.; Ye, Z. S.; Wang, D. S.; Chen, Q. A.; Zhou, Y. G. (2011). "An Enantioselective Approach to 2,3-Disubstituted Indolines through Consecutive Brønsted Acid/Pd-Complex-Promoted Tandem Reactions". Chemistry: A European Journal. 17 (26): 7193–7. doi:10.1002/chem.201100576. PMID21567504.
↑Wang, D. S.; Ye, Z. S.; Chen, Q. A.; Zhou, Y. G.; Yu, C. B.; Fan, H. J.; Duan, Y. (2011). "Highly Enantioselective Partial Hydrogenation of Simple Pyrroles: A Facile Access to Chiral 1-Pyrrolines". Journal of the American Chemical Society. 133 (23): 8866–8869. Bibcode:2011JAChS.133.8866W. doi:10.1021/ja203190t. PMID21591641.
↑Wang, D. S.; Chen, Q. A.; Lu, S. M.; Zhou, Y. G. (2012). "Asymmetric Hydrogenation of Heteroarenes and Arenes". Chemical Reviews. 112 (4): 2557–2590. doi:10.1021/cr200328h. PMID22098109.
↑Urban, S.; Beiring, B.; Ortega, N.; Paul, D.; Glorius, F. (2012). "Asymmetric Hydrogenation of Thiophenes and Benzothiophenes". Journal of the American Chemical Society. 134 (37): 15241–15244. Bibcode:2012JAChS.13415241U. doi:10.1021/ja306622y. PMID22934527.
12Heitbaum, M.; Glorius, F.; Escher, I. (2006). "Asymmetric Heterogeneous Catalysis". Angewandte Chemie International Edition. 45 (29): 4732–62. doi:10.1002/anie.200504212. PMID16802397.
↑Yoon, M.; Srirambalaji, R.; Kim, K. (2012). "Homochiral Metal–Organic Frameworks for Asymmetric Heterogeneous Catalysis". Chemical Reviews. 112 (2): 1196–1231. doi:10.1021/cr2003147. PMID22084838.
↑Hu, A.; Ngo, H. L.; Lin, W. (2003). "Chiral Porous Hybrid Solids for Practical Heterogeneous Asymmetric Hydrogenation of Aromatic Ketones". Journal of the American Chemical Society. 125 (38): 11490–11491. Bibcode:2003JAChS.12511490H. doi:10.1021/ja0348344. PMID13129339.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.